RESULTS AND DISCUSSION
From the downcore variation of dissolved sulfate concentrations in the interstitial waters (Fig. 2), it is evident that most of the sites are characterized by more or less intense bacterial sulfate reduction. Even at those sites where the pore-water sulfate concentrations are comparable to those of modern Mediterranean seawater (~31 mM; surface seawater at Site 973; Böttcher et al., 1998), microbial activity using sulfate as the electron acceptor is observed from the sulfur isotopic trends of residual sulfate (see section on sulfur isotopes).
The reduction of dissolved sulfate is coupled to the availability of metabolizable organic matter in the sediments. Whereas sulfate reduction is essentially complete in the upper meters of Sites 976 and 977 (Table 1; Fig. 2), significant amounts of sulfate are present in the interstitial waters of all other sites. No influence of organic-rich layers (sapropels) in the sediment column on the pore-water sulfate profiles is observed. Sulfate reduction rates for the upper parts of the sediment sections of all sites except for 978 were calculated according to Canfield (1991) and are positively correlated to the bulk sedimentation rates (Fig. 3), which is consistent with higher preservation of metabolizable organic matter with increasing sedimentation rate (Berner, 1980). The results for the western Mediterranean (Leg 161; this study) compare well with those for the eastern Mediterranean (Leg 160; Böttcher et al., 1998). It should be noted that the sulfate reduction rates were determined by modeling the sulfate profiles (Canfield, 1991), which may depart from direct measurements derived from 35SO42- incubations.
The sulfate profiles at Sites 976 and 977, and probably Site 979, show a convex-up curvature (Fig. 2) which, together with the downward increase in alkalinity and dissolved ammonium (Comas, Zahn, Klaus, et al., 1996), indicate that sulfate reduction seems to be related to the microbial in situ degradation of organic matter and that upward diffusion of methane played no significant role in the upper part of the sedimentary column (Borowski et al., 1996). For Sites 974, 975, and 978, an increase in sulfate concentrations is found deeper downcore (Fig. 2). This is caused by a superimposition of bacterial sulfate reduction by a sulfate input from the dissolution of upper Miocene evaporites or the influence of saline brines located at depth. The upward sulfate flux from evaporitic brines is inferred from salinity and major-element variations of the interstitial waters from Sites 974 and 978 (Comas, Zahn, Klaus, et al., 1996). These brines may be paleofluid that had been trapped below the Pliocene-Pleistocene sediments or brines derived from salt dissolution (Comas, Zahn, Klaus, et al., 1996). Evaporite dissolution may have occurred in underlying strata or in distant Messinian salt deposits followed by large-scale fluid migration along permeable sediments to the investigated site (Comas, Zahn, Klaus, et al., 1996). At Sites 975 and 978, gypsum was found in the deeper sediment cores and the pore-water profiles provide evidence for the dissolution of calcium sulfates (Comas, Zahn, Klaus, et al., 1996;
Bernasconi, Chap. 33, this volume).
The microbial reduction of dissolved sulfate leads to a kinetic isotope effect and an enrichment of the lighter sulfur isotope, 32S, in the formed hydrogen sulfide and a corresponding increase in the isotope composition of the residual sulfate (e.g., Chambers and Trudinger, 1979). Rayleigh fractionation evaluation of the residual sulfate data of Sites 975, 976, and 977, assuming closed system conditions with respect to dissolved sulfate (Hartmann and Nielsen, 1969; Sweeney and Kaplan, 1980), yields linear correlations (Fig. 4) and fractionation factors between 1.019 and 1.066. The values for Sites 976 and 977 are within the range observed experimentally for microbial sulfate reduction at low reaction rates (Chambers and Trudinger, 1979; Canfield and Teske, 1996; Rees, 1973). A contribution of sulfate diffusion from the sediment-water interface (i.e., SO42- that is 34S depleted relative to the residual pore-water sulfate) to the interstitial water sulfate pool would increase the calculated fractionation factors (Jørgensen, 1979). The calculated fractionation factor for the upper sediment column at Site 975 exceeds the experimentally observed range, which may be caused by a very low sulfate reduction rate.
A superimposition of bacterial sulfate reduction is found in the samples from Site 975 below ~47 meters below seafloor (mbsf) by a sulfate input from upper Miocene evaporites located at depth with a 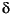 34S value ~+23
34S value ~+23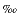 (Table 2). The dissolution of sulfate minerals from evaporites does not lead to sulfur isotope fractionation (Böttcher and Usdowski, 1993). Therefore, late Miocene sulfates contribute to the sulfur isotopic composition of the interstitial sulfate with a 34S value of ~+23, in general agreement with the observed variation of 34S values at Site 975 (Fig. 2). Considering an enrichment of 34S by ~+1.6 in the solid during crystallization of gypsum (Thode and Monster, 1965), the parent solution of Site 975 gypsum should have had an isotopic composition ~+21, which is similar to that of modern Mediterranean seawater (Böttcher et al., 1998; De Lange et al., 1990), and the inferred variation of seawater isotopic composition within the last 10 Ma (Burdett et al., 1989). The sulfur isotopic composition of dissolved sulfate at 576 mbsf at Site 978 (+27.1; Table 1) demonstrates that Messinian calcium sulfates and/or saline brines were probably the source of the dissolved sulfate, and that minor microbial sulfate reduction occurred. Trapped late-stage evaporitic brines or brines derived from the dissolution of Messinian salts should contribute sulfate with a 34S value ~+23.
(Table 2). The dissolution of sulfate minerals from evaporites does not lead to sulfur isotope fractionation (Böttcher and Usdowski, 1993). Therefore, late Miocene sulfates contribute to the sulfur isotopic composition of the interstitial sulfate with a 34S value of ~+23, in general agreement with the observed variation of 34S values at Site 975 (Fig. 2). Considering an enrichment of 34S by ~+1.6 in the solid during crystallization of gypsum (Thode and Monster, 1965), the parent solution of Site 975 gypsum should have had an isotopic composition ~+21, which is similar to that of modern Mediterranean seawater (Böttcher et al., 1998; De Lange et al., 1990), and the inferred variation of seawater isotopic composition within the last 10 Ma (Burdett et al., 1989). The sulfur isotopic composition of dissolved sulfate at 576 mbsf at Site 978 (+27.1; Table 1) demonstrates that Messinian calcium sulfates and/or saline brines were probably the source of the dissolved sulfate, and that minor microbial sulfate reduction occurred. Trapped late-stage evaporitic brines or brines derived from the dissolution of Messinian salts should contribute sulfate with a 34S value ~+23.
Figure 5 summarizes all measured sulfur isotope data as a function of the residual sulfate concentration and compares the results with some predicted general trends, which are, alone or in combination, responsible for the observed variations in the interstitial waters from Leg 161. From a comparison of the analyzed pore waters with these trends, it is evident that the dominant processes influencing the relationships between concentration and sulfur isotopic composition of residual dissolved sulfate are microbial sulfate reduction and sulfate derived from Messinian calcium sulfates.
The oxygen isotopic composition of residual sulfate was measured for selected interstitial waters from Sites 974, 975, 977, and 979 (Table 3). The dissolved sulfate was generally enriched in 18O with respect to modern Mediterranean seawater sulfate (18O[SO42-] 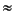 +9.4; Cortecci, 1974a, 1974b), and the values of 18O(SO42-) increase with depth (Fig. 6). Although, the 18O(SO42-) values increase with increasing 34S values, the oxygen isotopic composition of the residual sulfate tends to approach an asymptotic value (Fig. 7). For the pore waters of the upper few tens of meters of the profiles, the 18O(H2O) values range between +1.1 and +1.5 (Table 2). Because of early diagenetic reactions, the values decrease slightly with depth. Water temperatures of 16°, 12°, 15°, and 11°C were measured at Sites 974, 975, 977, and 979 at ~22 mbsf (Comas, Zahn, Klaus, et al., 1996) and increased further downcore. At ~50 mbsf, for instance, temperatures of 17° (Site 977) and 14°C (Site 979) were found, respectively (Comas, Zahn, Klaus, et al., 1996). Using 15°C as a typical water temperature in the sediment sections analyzed for 18O(SO42-), the isotopic composition of dissolved sulfate in equilibrium with interstitial waters of +1.3 composition should be +32.4 when compared to the extrapolated results from hydrothermal inorganic exchange experiments (Mizutani and Rafter, 1969). Consideration of a range of temperatures between 10° and 20°C leads to theoretical boundary values for the equilibrium isotope composition of dissolved sulfate of +31.2 and +33.7 (Mizutani and Rafter, 1969). The observed stable isotope data are generally below the proposed equilibrium value (Fig. 6, Fig. 7).
+9.4; Cortecci, 1974a, 1974b), and the values of 18O(SO42-) increase with depth (Fig. 6). Although, the 18O(SO42-) values increase with increasing 34S values, the oxygen isotopic composition of the residual sulfate tends to approach an asymptotic value (Fig. 7). For the pore waters of the upper few tens of meters of the profiles, the 18O(H2O) values range between +1.1 and +1.5 (Table 2). Because of early diagenetic reactions, the values decrease slightly with depth. Water temperatures of 16°, 12°, 15°, and 11°C were measured at Sites 974, 975, 977, and 979 at ~22 mbsf (Comas, Zahn, Klaus, et al., 1996) and increased further downcore. At ~50 mbsf, for instance, temperatures of 17° (Site 977) and 14°C (Site 979) were found, respectively (Comas, Zahn, Klaus, et al., 1996). Using 15°C as a typical water temperature in the sediment sections analyzed for 18O(SO42-), the isotopic composition of dissolved sulfate in equilibrium with interstitial waters of +1.3 composition should be +32.4 when compared to the extrapolated results from hydrothermal inorganic exchange experiments (Mizutani and Rafter, 1969). Consideration of a range of temperatures between 10° and 20°C leads to theoretical boundary values for the equilibrium isotope composition of dissolved sulfate of +31.2 and +33.7 (Mizutani and Rafter, 1969). The observed stable isotope data are generally below the proposed equilibrium value (Fig. 6, Fig. 7).
The inorganic oxygen isotope exchange reaction between dissolved sulfate and water at low temperatures and neutral pH is extremely slow (e.g., Chiba and Sakai, 1985; Mizutani and Rafter, 1969), and has been found to be negligible in oxic deep-sea sediments up to 50 m.y. old (Zak et al., 1980). However, significant oxygen isotope variations in dissolved sulfate have been observed in microbial sulfate reduction studies (e.g., Mizutani and Rafter, 1973; Fritz et al., 1989) and pore waters of anoxic sediments (Zak et al., 1980; Böttcher et al., 1998; M. E. Böttcher, unpubl. data). In the initial stage of sulfate reduction, kinetic isotope effects are expected to be responsible for a common increase in 18O and 34S values because the 32S-16O bonds are weaker than the 34S-16O and 32S-18O bonds (Zak et al., 1980). From Figure 4, it is evident that the variation of 18O(SO42-) values as a function of ln F does not fall on a linear trend, as expected for an unidirectional kinetic isotope fractionation. As outlined by Böttcher et al. (1998), this difference is caused mainly by oxygen isotope exchange reactions with the aqueous solution via a sulfate-enzyme complex, which is formed as an intermediate reaction product (Fritz et al., 1989), leading to an increased equilibration between residual sulfate and pore water with increasing degree of microbial sulfate reduction. Figure 7 shows that the oxygen isotope data of pore-water sulfate increase more rapidly than the 34S values increase at Sites 974 and 975 compared to Site 977. This is caused by lower sulfate reduction rates at Sites 974 and 975 (Fig. 3), which enables a more intense oxygen isotope exchange upon reaction even at low degrees of sulfate reduced. The distinct relationships between 18O and 34S values seem to be related to their different sulfate reduction rates, thus confirming the previous suggestion of Böttcher et al. (1998) that different sulfate reduction rates in marine sediments are directly reflected by 18O-34S plots.
The decrease in 18O(SO42-) at greater depth at Site 975 (Fig. 6) results from the dissolution of upper Miocene evaporites with an approximate oxygen isotope value of +16 (Stenni and Longinelli, 1990). The slight depletion of the interstitial water in 18O at greater depth, which is caused by some diagenetic water-rock interactions
(Bernasconi, Chap. 33, this volume), should only have been a minor influence on the composition of dissolved sulfate. The oxygen isotope data of dissolved sulfate for Site 977 level off below the theoretical equilibrium value of +32.4. This observation is similar to the experimental results of Fritz et al. (1989), in which a smaller isotope fractionation was observed at steady state probably results from an uncertainty in the values.
The carbon isotopic compositions of dissolved inorganic carbonate species at Sites 974, 975, 976, 977, and 979 vary between -0.1 and -22.6 relative to V-PDB (Table 1). The downcore profiles of all sites show similar trends. Specifically, in the uppermost part of the sediments, at depths at or above 50 mbsf, observed minima in the 13C records probably result from the microbial degradation of organic matter by sulfate-reducing bacteria and the concomitant liberation of CO2. The extent of 13C depletion in dissolved inorganic carbonate species at the minima is directly related to the amount of sulfate reduced (Fig. 2, Fig. 8). The subsequent increase in 13C of dissolved inorganic carbonate species with increasing depth at Sites 976, 977, and 979 is caused by bacterial methanogenesis, which succeeds sulfate reduction in anaerobic organic carbon-rich sediments and is clearly reflected by the variation of measured headspace methane concentrations (Fig. 8). Because the activity of methane-producing bacteria leads to the formation of methane enriched in 12C (Games et al., 1978; Carothers and Kharaka, 1980; Botz et al., 1997), the interstitial water can become enriched in 13C, and the pore waters evolve to higher 13C values as bacterial methane formation continues.
The decrease of 13C in the deepest intervals of Sites 977 and 979 (Fig. 8) is probably related to a repeated onset of microbial activity oxidizing organic matter or methane. At least for Site 977, the increased availability of dissolved sulfate indicates that sulfate may act again as the electron acceptor. Probably, the lowermost pore-water sample analyzed at Site 979 became completely depleted in sulfate because of microbial sulfate reduction, and sulfate is expected to increase in the underlying sediments.
It should be noted, however, that the variations of dissolved calcium and magnesium (Comas, Zahn, Klaus, et al., 1996;
Bernasconi, Chap. 33, this volume) and alkalinity with depth (Fig. 8) also provide evidence for an influence on the isotopic composition of dissolved inorganic carbonate species by carbonate dissolution and precipitation reactions. The downcore increase of 13C values at Sites 974 and 975 can only be related to carbonate diagenesis, because the high dissolved sulfate concentrations hindered methanogenesis (e.g., Carothers and Kharaka, 1980).
