Selected 1.5-m sections of intact core from Holes 994B and 995B were cut from the standard 9-m segments, capped immediately, and transferred to a cold room (6ºC) for storage. The sections were placed into PVC tubes that were capped on both ends and fitted with gas inlet and outlet ports. These ports allowed for continuous flushing with nitrogen gas to ensure anoxia of the cores during storage.
Cores from Hole 994B were subsampled aboard the JOIDES Resolution approximately one week after they were obtained. Cores from Hole 995B were transported to a shore-based lab and subsampled approximately two weeks after they were obtained. Subsampling of the cores was conducted in similar fashion for both ship- and shore-based measurements. Desired depth ranges were sampled by hack sawing through the core liner and sediment (perpendicular to the axis of the core). The selected intervals were at least 10 cm from either end of the original core section so that relatively undisturbed samples were assured. Immediately after sawing, the freshly exposed sediment surface was sampled by inserting cut-off 3-mL plastic syringes along the axis of the core (thus sampling a depth interval of ~4 cm). The syringe plunger was held fixed at the sediment surface while the barrel was pushed into the sediment, in an overall procedure that is analogous to piston coring. This sampling method produced sediment-filled syringes that were free from atmospheric gas bubbles. The syringes were then removed from the sediment and treated as described below for the concentration/isotope and rate measurements.
Pore-water methane gas was sampled by advancing the plunger of the open-ended plastic syringe to extrude a small quantity of sediment, then shaving off this excess (with a razor blade) so that the surface of the remaining sediment was flush with the open end of the syringe barrel. This allowed for precise determination of the sediment volume contained in the (graduated) syringe. The sediment in the syringe was then extruded into a 20-mL glass serum vial that contained 5.00 mL of 1.0 M NaOH (designed to terminate any bacterial activity). The vial was then capped immediately with a red rubber septum. The internal volume of each sample vial was carefully determined and recorded beforehand, taking into account the effect of the rubber septum.
Each vial was shaken vigorously for several minutes and then left to stand for >1 hr. This process slurried the sediment pellet into a high-porosity suspension that promoted equilibration of pore-water methane with the vial headspace. We have previously found that suspensions of similar porosity achieve aqueous-gas equilibrium within minutes. The headspace gas was sampled by syringe: a small volume (1 mL or less) of distilled water was injected into the sealed vial and an equivalent volume of gas was withdrawn. The methane concentration in the headspace gas was analyzed by gas chromatography with flame ionization detection. Standards were prepared minutes before use by diluting precisely known volumes of pure methane gas into glass serum vials similar to those used for the sediment samples. The pore-water methane concentration was then calculated from the headspace gas concentration as follows:
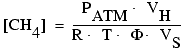 ,
,
where VH and VS
are the volume of the sample vial headspace and whole sediment sample,
respectively; PATM is the pressure of the vial headspace (assumed to
be the measured atmospheric pressure at the time the vials were sealed); R is
the universal gas constant; T is the temperature of the vial headspace; and ![]() is the sediment porosity (determined in our lab for three depth intervals over
the sampled length of core).
is the sediment porosity (determined in our lab for three depth intervals over
the sampled length of core).
Once the vials had been sampled for methane concentration analysis, they were subsequently sampled for stable carbon isotope analysis. Distilled water (5 mL) was injected into the vial by syringe and an identical volume of headspace gas was removed. Methane in the sample was concentrated in-line via cryogenic focusing (Popp et al., 1995). The 13C/12C ratio was determined on a Mat-252 isotope ratio mass spectrometer (Finnigan Mat). Reported isotope ratios are corrected for the contribution of atmospheric methane in the headspace gas to the overall methane isotope signature. The isotopic composition of atmospheric methane was measured for "lab air" at the time the sample vials were sealed.
For rate measurements, the sediment-filled syringes were sealed with black-butyl rubber stoppers. These stoppers had one hole that had previously been filled with silicone caulk to provide a septum for injecting tracer. The filled syringes were incubated overnight in a constant temperature bath at the in situ sediment temperature. Each syringe was then injected with an aliquot of either aqueous H235SO4 (specific activity = 1500 Ci·mmol-1; 10-µL injection; 3 × 108 dpm for samples in the 0-17 mbsf interval or 3 x 107 dpm for those below 17 mbsf) or gaseous 14CH4 (specific activity = 55 mCi·mmol-1; 25-µL injection; 7 x 106 dpm). In the case of 35S, the added sulfur was negligible with respect to the ambient pool. The 14C addition enhanced the ambient CH4 concentration by ~3 µM (equivalent to a 30% increase at the top of the sediment column and a 1%-5% increase in the apparent zone of methane oxidation). The enhanced methane concentration was taken into account in all rate calculations. The injected samples were incubated for 4 days at in situ sediment temperature in the dark. Incubations were terminated by extruding the sediment pellet into a 20-mL glass serum vial containing 5.0 mL of 1.0 M NaOH and immediately capping and shaking the vial.
The 35S tracer was obtained as H235SO4 from a commercial source and used as is. Because there was no commercially available source of 14CH4 at the time of cruise preparations, tracer was obtained as 14CH3I and reacted to methane. This was accomplished by adding an excess of Superhydride (Aldrich Chemical Co.) to the CH3I under anhydrous and anoxic conditions. Reductive dehalogenation of the CH3I produced methane, which was transferred to a vial containing an anoxic solution of 0.1 M NaOH (ensuring that any 14CO2 present would be removed from the tracer gas). Significant dilution of the methane with nitrogen gas occurred during the overall process, such that the activity of the resultant tracer gas was limited to 7 x 106 dpm per 25-µL injection. The synthesized gas was free of 14CO (by GC/HgO detection) and 14C2 or higher hydrocarbons (by GC/FID), ensuring that any 14CO2 formed during the sediment incubations must have derived entirely from 14CH4.
For the sulfate reduction rate samples, the quantity of H235S formed during the incubation was measured by a previously described acidic-chrome technique (Fossing and Jørgensen, 1989). This was used to calculate sulfate reduction rates (SRR) as follows:
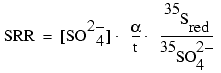 ,
,
where ![]() is the kinetic isotope fractionation factor (35S vs. 32S)
observed for sulfate reduction (assumed to be 1.045; Jørgensen, 1978); t is the
incubation time; 35Sred is the recovered activity of
reduced sulfur forms; and 35SO42- is the added
activity of 35SO42- .
is the kinetic isotope fractionation factor (35S vs. 32S)
observed for sulfate reduction (assumed to be 1.045; Jørgensen, 1978); t is the
incubation time; 35Sred is the recovered activity of
reduced sulfur forms; and 35SO42- is the added
activity of 35SO42- .
During the initial stages of our analysis, the acidic-chrome technique was found to produce a slow but constant chemical reduction of the tracer 35SO42- during sample analysis, thus contributing to a high and somewhat variable blank. This chemical reduction is made problematic by the necessity to measure sulfate reduction rates that are 2-4 orders of magnitude lower than those typically observed in coastal sediments. To address this problem one of the two replicate samples from each depth was treated with a procedure intended to remove tracer 35SO42- prior to analysis. The sediment sample was slurried with a large quantity of distilled water (~50 mL), allowed to settle for several hours, and then decanted (retaining the solid phase). This procedure was repeated, and the solid phase was then subjected to the acidic-chrome procedure. Because of the potential for loss of reduced sulfur during the above described procedure, these values are probably best viewed as minimum estimates. This potential loss, combined with possible blank-induced variability in the nondecanted samples contributes to considerable noise between replicates. Despite this noise, relative depth-variations in sulfate reduction rates can be clearly discerned.
For methane oxidation rate samples, the quantity of 14CO2 and 14CH4 in each sample at the end of the incubation was determined via the method described in Hoehler et al. (1994). These values were used to calculate methane oxidation rates (MOR) as follows:
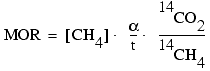 ,
,
where ![]() is the kinetic isotope fractionation factor (14C vs. 12C)
observed for methane oxidation (assumed to be 1.02; Alperin et al., 1988); t is
the incubation time; and 14CO2 and 14CH4
are recovered radioactivities.
is the kinetic isotope fractionation factor (14C vs. 12C)
observed for methane oxidation (assumed to be 1.02; Alperin et al., 1988); t is
the incubation time; and 14CO2 and 14CH4
are recovered radioactivities.
![]()