FROM OXIDIZING TO REDUCING ALTERATION
The transition from alteration processes controlled by the presence of fractures to those related to the pervasive fluid seepage within the unfractured rock separates two different styles of alteration that respectively occur under oxidizing and reducing conditions. Although several exceptions exist, the most common mineral paragenesis characterizing the two styles of alteration are quite distinct and are in good agreement with those observed by numerous authors (e.g., Andrews 1977, 1980; Alt, Kinoshita, Stokking, et al., 1993; Laverne et al., 1996; Teagle et al., 1996): (1) assemblages under oxidizing conditions: Fe-oxyhydroxides ± iddingsite ± celadonite ± Mg-rich saponite; and (2) assemblages under reducing conditions: Fe-rich saponites ± carbonates ± sulfides ± talc.
Sensitive markers of the transition from assemblages 1 to 2 are the mineralogical and minerochemical variations involving the clay minerals, the mixed-layer clays, and the clay mixtures. As evidenced in
Figure 14 (where the complete set of the analyzed secondary minerals is reported), good indicators of the major chemical variations are the K2O, FeOt, and MgO contents of these secondary phases. Following the crystallization order in the polymineralic fillings of the gas vesicles from the different zones discussed in the previous section, it is possible to associate these chemical changes with the spatial and temporal variations occurring during the alteration processes.
As evidenced in Figure 15 (Steps 1-2) the most important variation occurring in the early stages of the oxidizing alteration (i.e., in the reddish brown and greenish halos) is the progressive decrease of Fe-rich minerals associated with the concomitant increase of K-bearing minerals and mixtures (i.e., Fe-oxyhydroxides 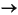 iddingsite iddingsite-celadonite mixtures celadonite-iddingsite mixtures celadonite). Oxygen isotope data suggest temperatures up to 40°C for the formation of celadonite and celadonite-bearing mixtures (Kastner and Gieskes, 1976; Seyfred et al., 1978; Andrews, 1980; Böhlke et al., 1984). Consequently, this assemblage forms at very low temperatures, slight alkaline conditions, high oxidation potentials, and requires substantial supplies of Fe and K from the circulating fluids. All these constraints suggest that seawater is the fluid responsible for this early alteration stage. Nevertheless, although K is indigenous to normal seawater, significant amounts of iron are required to justify the massive precipitation of Fe-oxyhydroxides and iddingsite. Presumably iron is supplied to the circulating fluids in part by the breakdown of olivine and in part during the reaction that occurred in the fracture systems of the adjacent glassy rims.
iddingsite iddingsite-celadonite mixtures celadonite-iddingsite mixtures celadonite). Oxygen isotope data suggest temperatures up to 40°C for the formation of celadonite and celadonite-bearing mixtures (Kastner and Gieskes, 1976; Seyfred et al., 1978; Andrews, 1980; Böhlke et al., 1984). Consequently, this assemblage forms at very low temperatures, slight alkaline conditions, high oxidation potentials, and requires substantial supplies of Fe and K from the circulating fluids. All these constraints suggest that seawater is the fluid responsible for this early alteration stage. Nevertheless, although K is indigenous to normal seawater, significant amounts of iron are required to justify the massive precipitation of Fe-oxyhydroxides and iddingsite. Presumably iron is supplied to the circulating fluids in part by the breakdown of olivine and in part during the reaction that occurred in the fracture systems of the adjacent glassy rims.
The next stage of the alteration (Steps 2-3; Fig.
15) is recorded by the appearance of the Mg-rich saponite as the dominant secondary mineral. As discussed by Andrews (1980) the transition from Fe- and K-rich assemblages to Mg-rich saponite reflects a gradual and systematic extraction of oxygen from the circulating fluids during the progressive interaction with the host rock. The progressive change of the fluid chemistry is associated with a gradual closure of the main circulation pathways, which represents the first step of the transition from a fluid-dominated to a rock-dominated system.
The first growth of Fe-rich saponites, often associated with scattered but widespread sulfides, and the contemporaneous disappearance of secondary minerals that require high oxidation potential (such iddingsite and celadonite) mark the transition to the reducing style of alteration (Steps 3-4;
Fig. 15). The chemical variations from Mg- to Fe-rich saponites that are associated with this transition reflect a new increased availability of ferrous iron in the circulating fluids. This is presumably related to a significant decrease of Eh of the environment (at least in the region of sulfide stability), which allows iron to be removed from the host rock and mobilized in the fluid.
Nevertheless the complete data set of clay analyses from the present study and from other members of the ODP Leg 168 Scientific Party (Hunter et al., 1999; Porter et al.,
Chap. 12, this volume) seem to indicate that the variation of Fe and Mg content in saponite could also be related to the bulk rock chemistry of the basalts. In fact all saponites (both Mg rich and Fe rich) from Site 1025 and subordinately from Site 1029 (i.e., those present in fractionated ferrobasalt) always show the lower Mg/Fet ratio when compared to the other occurrences
(Fig. 7).
The last stage of the alteration begins with the first appearance of carbonates (Step 4-5;
Fig. 15); this stage has been recognized only in samples from Sites 1025, 1027, 1028, and 1032. At the beginning an almost pure CaCO3 carbonate (in most of the cases aragonite; Yatabe et al.,
Chap. 11, this volume) clearly coprecipitated together with variable proportions of Fe-rich saponites in the outer rims of the vein. Subsequently, calcite with significant amounts of Mn and minor Fe and Mg became the dominant secondary phase. A third generation of carbonates (almost pure CaCO3 calcite) has been recognized only in samples from Sites 1027 and 1032. This evidence suggests that while carbonates precipitated together with saponite, their dominant cation is Ca2+ with other cations being partitioned into the clay minerals. As the saponite became progressively a minor constituent, the composition of carbonates changed to include Mn2+, Fe2+, and Mg2+, along with Ca2+. The source of bicarbonate necessary to achieve carbonate saturation in the fluid is not completely clear. One possible source is dissolution within the base of turbiditic sediments onlapping the pillow basalts. If this is so, then carbonates and some other phases related to the reducing stages of alteration occurred when the eastern flank of the JdFR became buried, and open circulation of seawater within the basement became restricted or absent.
Hunter et al. (1999) suggested temperatures of formation for the carbonates varying from about 35° to 70°C. As for the oxidizing stages of the alteration, these temperatures are in good agreement with those estimated at the present-day basement/sediment interface (Davis, Fisher, Firth, et al., 1997).
