ANALYTICAL METHODS
All samples were washed with water over a 0.5-mm mesh sieve to remove the mud fraction, then dried in an oven at 25°C. Fragments of bryozoan skeletons in a good state of preservation were selectively picked under a binocular microscope. Before mineralogical and isotopic analyses, the skeletons were broken into several pieces and the fine carbonate fractions were removed under a binocular microscope. Several skeletons were observed with a field emission scanning electron microscope (SEM) (JEOL JSM-6330F) or with a low-vacuum SEM (JEOL JSM-5800LV) to check whether the material had been diagenetically altered. Although a small amount of carbonate cement was observed in the skeletons collected at depths >40 mbsf (Fig. F6), further pretreatment to remove cements such as hydrogen peroxide soaking (Sakai and Kano, 2001) was not carried out because our preliminary analysis indicates that the H2O2 treatment affected the isotopes. All samples were powdered prior to mineralogical analyses.
Mineralogy and molar percentage (mol%) MgCO3 of bryozoan of skeletal carbonate were determined by X-ray diffraction (XRD) using a Philips X'pert-MPD (PW3050) with CuK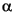 radiation and a Ni filter. Samples (~0.5-1.0 mg) were ground to fine particles (~10 µm in diameter) by careful grinding by hand. A smear mount was prepared by smearing the particles with distilled water on to a silicon plate (nonreflection) and then air dried. The goniometer scanned from 20° to 40°2
radiation and a Ni filter. Samples (~0.5-1.0 mg) were ground to fine particles (~10 µm in diameter) by careful grinding by hand. A smear mount was prepared by smearing the particles with distilled water on to a silicon plate (nonreflection) and then air dried. The goniometer scanned from 20° to 40°2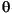 with a step size of 0.02°2 and a count time of 2.0 s/step for all samples. A tube voltage of 40 kV and a tube current of 55 mA were used. Diffractograms were stripped to remove K2 signal and smoothed. Mineral weight percentages were calculated by the integrated peak intensity procedures following Cook et al. (1975). Magnesium content in skeletal carbonate was determined by displacement of the d104 peak of calcite with increasing MgCO3 (Goldsmith and Graf, 1958). Precision of XRD through the whole experiment was estimated by measuring the positions of the d111 and d220 peaks of silicon before and after a sequences of daily analyses (15-20 samples) and was less than ±0.01°2. This implies that concentrations determined by XRD are accurate to ±0.3 mol% MgCO3.
with a step size of 0.02°2 and a count time of 2.0 s/step for all samples. A tube voltage of 40 kV and a tube current of 55 mA were used. Diffractograms were stripped to remove K2 signal and smoothed. Mineral weight percentages were calculated by the integrated peak intensity procedures following Cook et al. (1975). Magnesium content in skeletal carbonate was determined by displacement of the d104 peak of calcite with increasing MgCO3 (Goldsmith and Graf, 1958). Precision of XRD through the whole experiment was estimated by measuring the positions of the d111 and d220 peaks of silicon before and after a sequences of daily analyses (15-20 samples) and was less than ±0.01°2. This implies that concentrations determined by XRD are accurate to ±0.3 mol% MgCO3.
The skeletons were processed by an automated carbonate device (Kiel III, Finnigan MAT GmbH) attached to a Finnigan MAT Delta S mass spectrometer at the Institute of Geology and Paleontology, Graduate School of Science, Tohoku University, or by the same device attached to a Finnigan MAT 252 mass spectrometer at the Technological Research Center, Japan National Oil Corporation (TRC/JNOC). A sample of ~0.1 mg was separately dissolved to CO2 by "105%" phosphoric acid in a vacuum at 70°C (at Tohoku University) or at 72°C (at TRC/JNOC) in a reaction vessel. The results of isotope analysis were corrected for the usual isobaric interferences using the equations of Santrock et al. (1985) and presented according to the conventional notation in parts per thousand deviation from the international standard, Vienna Peedee belemnite (VPDB). After correction for the difference in oxygen isotopic fractionation between reaction temperatures of 25°, 70°, and 72°C, 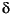 18O of carbonate sample was calculated using = 1.01025 at 25°C for calcite (Sharma and Clayton, 1965; Friedman and O'Neil, 1977) and = 1.01034 at 25°C for aragonite (Sharma and Clayton, 1965; Friedman and O'Neil, 1977). The conversion between VPDB and Vienna Standard Mean Ocean Water (VSMOW) scales was calculated using the equations of Coplen et al. (1983). External precision through the whole experiment was estimated from four to eight analyses of internal laboratory calcite standard (MACS1) inserted in every batch of samples (20-44 samples) and was <±0.05
18O of carbonate sample was calculated using = 1.01025 at 25°C for calcite (Sharma and Clayton, 1965; Friedman and O'Neil, 1977) and = 1.01034 at 25°C for aragonite (Sharma and Clayton, 1965; Friedman and O'Neil, 1977). The conversion between VPDB and Vienna Standard Mean Ocean Water (VSMOW) scales was calculated using the equations of Coplen et al. (1983). External precision through the whole experiment was estimated from four to eight analyses of internal laboratory calcite standard (MACS1) inserted in every batch of samples (20-44 samples) and was <±0.05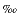 for 13C and ±0.08 for 18O (±1
for 13C and ±0.08 for 18O (±1 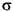 , N = 72). Several measurements of the internal laboratory standards showed that differences in isotopic compositions of the same material between two laboratories was negligibly small (<0.1 for 13C and 18O).
, N = 72). Several measurements of the internal laboratory standards showed that differences in isotopic compositions of the same material between two laboratories was negligibly small (<0.1 for 13C and 18O).
