MATERIALS AND METHODS
Sample Treatment, Analytical Methods, and Data Quality
Many mixed-layer phyllosilicates become unstable at temperatures of ~100°C, resulting in phase transformation and loss of volatile components (mainly H2O or OH) contained in the crystal lattice. Because mixed-layer phyllosilicates are prominent in the hydrothermally altered dacites from the PACMANUS system, the powders were dried at 80°C overnight prior to any analyses.
Major elements and some trace elements (Ba, Rb, Sr, Y, and Zr) were measured by X-ray fluorescence (XRF) using fused disks at the Mineralogisch-Petrologisches Institut (Universität Bonn, Germany). The internal standard powders from Holes 1188B and 1191A were included in three batches, and the results of these measurements on individual fused disks provide an indication of the reproducibility of the data (Table T1). The analyses were performed on a Philips PW1480 X-ray spectrometer using Oxiquant software and 85 international standards for calibration. The totals for major elements and volatiles range from 98.5 to 101.5 wt%. In this regard, it was important to take the appropriate concentrations of water-soluble SO4 for anhydrite-rich samples into account.
Inductively coupled plasma–mass spectrometer (ICP-MS) measurements for Cu, Pb, Zn, Ba, Ag, As, Bi, Cd, Cr, Co, Ga, Mo, Ni, Rb, Sb, Sc, Sr, Tl, and U were performed on a PerkinElmer Elan 5000 instrument at the Institut für Mineralogie (Bergakademie Freiberg, Germany). Dissolution of the samples was achieved using concentrated HF and HNO3 under atmospheric conditions. However, for some samples, this method was not sufficient to completely dissolve the zircon microcrystals, which led to deficiencies in Zr, Hf, Th, Y, and heavy rare earth elements (HREE). This could be clearly recognized by examining the REE patterns and comparing the Zr measurements by ICP-MS to the Zr concentrations determined by XRF. Consequently, additional ICP-MS data were obtained from the ICP-MS laboratories at the University of Kiel and at the GeoForschungsZentrum Potsdam (GFZ). At GFZ Potsdam, samples were dissolved using HF, HClO4, and HCl in a multistage procedure that involved heating the samples at 180°C in pressurized Teflon vessels (Dulski, 2001). The solutions were analyzed with an Elan 5000A PerkinElmer Sciex instrument. At Kiel, glass beads were generated using ~250 mg of pulverized sample and lithium metaborate fusion. The beads were dissolved in HNO3, and the solutions were analyzed with an Agilent 7500c ICP-MS instrument. Details of this procedure are documented in Garbe-Schönberg (1993). The analyses presented in Table T1 have ZrXRF/ZrICP-MS ratios ranging from 0.8 to 1.2, which is an indication that the zircon microcrystals have been successfully dissolved and that the ICP-MS measurements of Zr, Hf, Th, Y, and HREE are accurate.
The H2O concentration was determined by thermal dissociation and infrared quantitative flow analysis using a Rosemount gas analyzer at the GEOMAR Institute (Kiel, Germany). Again, all samples were dried at 80°C to prevent destruction of some secondary phyllosilicates. Hence, values for the unaltered samples may include some water related to incipient hydration of the glassy groundmass that was not expelled at this temperature.
Measurements of S, C, and N were performed by thermal dissociation and element analysis in a VARIO EL gas analyzer (Institut für Mineralogie, Bergakademie Freiberg, Germany). Accuracy and precision were ensured by analyses of several duplicates and internal laboratory standards. For samples with >1 wt% Stotal, the content of water-soluble SO4 was determined gravimetrically at the Institut für Mineralogie (Bergakademie Freiberg, Germany) in order to account for the anhydrite content of many hydrothermally altered samples. About 2 g of sample powder was washed in ~500 mL of cold, distilled H2O for >12 hr. The dissolved SO4 was reacted with BaCl, causing quantitative precipitation of BaSO4, proportional to the concentration of the water-soluble sulfate in the sample. The detection limit is on the order of 0.6 wt% CaSO4 in the sample, and repeated analyses show that the relative error of the measurements is on the order of 5%.
The concentrations of F and Cl were determined for several samples at the Institut für Mineralogie (Bergakademie Freiberg, Germany) following procedures outlined by Klemm (1980). In order to remove any salt crystals (halite) from the samples, the samples were washed in warm water (30°C) for >1 hr prior to measurement. These powders were reacted with ZnO and Na2CO3 at 900°C, which liberated F and Cl from the silicate phases. These powders were dissolved in distilled H2O, and concentrations were determined using appropriate ion-selective electrodes. The detection limit was 200 ppm, and repeated analyses of particular samples indicate a relative error of 5% for F and Cl. A subset of seven samples was also analyzed for Cl at AnalytikJena AG (Germany) using a newly developed elemental analyzer (multi EA 2000), and these data are in agreement with our results (Table T1).
The 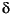 18Osilicate relative to the Vienna standard mean ocean water (VSMOW) was determined for 38 samples at the Mineralogisch-Petrologisches Institut (Universität Bonn, Germany). Where present, anhydrite was removed from the powders prior to analysis and the oxygen was liberated in a standard fluorination line using ultrapure F2 gas reacting with the sample powder at 600°C in Ni ovens. The liberated oxygen was transformed to CO2, and the 18Osilicate was measured with a SIRA-9 mass spectrometer. At least two individual 18Osilicate determinations were performed, and the average of these measurements is reported in Table T1. Deviation from the average is generally less than ±0.1
18Osilicate relative to the Vienna standard mean ocean water (VSMOW) was determined for 38 samples at the Mineralogisch-Petrologisches Institut (Universität Bonn, Germany). Where present, anhydrite was removed from the powders prior to analysis and the oxygen was liberated in a standard fluorination line using ultrapure F2 gas reacting with the sample powder at 600°C in Ni ovens. The liberated oxygen was transformed to CO2, and the 18Osilicate was measured with a SIRA-9 mass spectrometer. At least two individual 18Osilicate determinations were performed, and the average of these measurements is reported in Table T1. Deviation from the average is generally less than ±0.1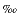 , but in some cases it is up to ±0.2.
, but in some cases it is up to ±0.2.
The 18Oanhydrite was determined for 24 samples using the BaSO4 powder generated during the gravimetric determination of water-soluble SO4. A method based on thermal combustion and online 18O measurement was applied at the Institut für Mineralogie (Bergakademie Freiberg, Germany) following procedures described by Kornexl et al. (1999). National Bureau of Standards (NBS) 127 reference material (BaSO4) was used for calibration of this method, and repeated analyses show that the reproducibility is generally ±0.3.
The 86Sr/87Srsilicate ratio was determined for 49 samples at the Institut für Mineralogie (Bergakademie Freiberg, Germany). Where present, anhydrite was removed to generate powders consisting exclusively of silicates. In addition, the 86Sr/87Sranhydrite ratio was measured for 24 samples that contain significant amounts of anhydrite (>1 wt% water-soluble SO4). The anhydrite was dissolved from the powders by washing in cold, distilled water for >12 hr. The measured 86Sr/87Sr ratio of the solutions is equal to the 86Sr/87Sranhydrite of the samples. All measurements were performed with a Finnigan MAT 263 mass spectrometer. Repeated analyses of 86Sr/87Srsilicate for individual samples were in excellent agreement and within the range of the error of the measurements (0.00001 to 0.00005). NBS 127 reference material was used to ensure accuracy of the measurements. Based on one duplicate analysis, the reproducibility of 86Sr/87Sranhydrite appears to be somewhat poorer (±0.0002), which could be related to problems during dissolution of the anhydrite from the sample powder.
The results of the mineral phase analysis by XRD are presented in Table T2. Step-scan XRD measurements (3°2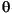 –80°2; 0.03°2 step size; and 5 s/step) were collected on spun samples with an RD 7 diffractometer (Seifert-FPM) at the Institut für Mineralogie (Bergakademie Freiberg, Germany). The diffractometer was equipped with a diffracted beam graphite monochromator and a variable divergence slit that allowed the irradiation of a constant sample area. A CuK
–80°2; 0.03°2 step size; and 5 s/step) were collected on spun samples with an RD 7 diffractometer (Seifert-FPM) at the Institut für Mineralogie (Bergakademie Freiberg, Germany). The diffractometer was equipped with a diffracted beam graphite monochromator and a variable divergence slit that allowed the irradiation of a constant sample area. A CuK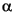 tube was used and operated at 40 kV and 40 mA. Qualitative phase analysis of the diffraction pattern was carried out by conventional search/match procedures using reference diffraction patterns stored in the International Centre for Diffraction Data (ICDD) PDF-2.
tube was used and operated at 40 kV and 40 mA. Qualitative phase analysis of the diffraction pattern was carried out by conventional search/match procedures using reference diffraction patterns stored in the International Centre for Diffraction Data (ICDD) PDF-2.
Using the results of the XRD analyses and the major element data, it was possible to run iterative calculations using the SOLVER function in an Excel spreadsheet to calculate normative mineral abundances. Based on assumed chemical compositions of the detected mineral phases, this calculation optimizes the proportions of the minerals until the calculated bulk rock composition is closest to the measured bulk rock composition. The results of these procedures are presented in Table T1.
