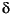 13C were poisoned with a saturated mercuric chloride solution (50 µL mercuric chloride solution to 1 mL interstitial water sample).
13C were poisoned with a saturated mercuric chloride solution (50 µL mercuric chloride solution to 1 mL interstitial water sample).
Shipboard interstitial water analyses were performed on 5-cm-long whole-round sections that were cut immediately after the core arrived on deck. In most cases, one whole-round section was taken from the middle of each core for the first six cores and every third core thereafter. To avoid the potential destruction of critical intervals, whole-round sections were not removed from cores adjacent to such intervals as determined by shipboard biostratigraphy. Details of the sampling resolution are described in the individual site chapters. After extrusion from the core liner, the surface of each whole-round section was scraped with a spatula to remove potential contamination. Interstitial waters were collected using a titanium squeezer, modified after the standard ODP stainless steel squeezer of Manheim and Sayles (1974). Pressure up to 205 MPa (30,000 psi) was applied using a hydraulic press. Pore waters were passed through prewashed Whatman No. 1 filters fitted above a titanium screen and subsequently extruded into a plastic syringe attached to the bottom of the squeezer assembly. All interstitial water samples were double-filtered through 0.45-µm polycarbonate filters. Samples for shipboard analysis were stored in plastic vials pending analysis. Aliquots for future shore-based analyses were placed in glass ampules and heat sealed. Aliquots for shore-based analysis of interstitial water 13C were poisoned with a saturated mercuric chloride solution (50 µL mercuric chloride solution to 1 mL interstitial water sample).
Interstitial water samples were routinely analyzed for salinity as total dissolved solids with a Goldberg optical handheld refractometer. The pH and alkalinity (Alk) were determined by Gran titration with a Brinkmann pH electrode and a Metrohm autotitrator. Dissolved chloride was determined by titration with silver chloride (AgCl). Sodium (Na+), potassium (K+), magnesium (Mg2+), calcium (Ca2+), and sulfate (SO42-) were analyzed by ion chromatography using a Dionex DX-120 ion chromatograph. Silica (Si[OH]4), phosphate (HPO42-), and ammonium (NH4+) concentrations were determined by spectrophotometric methods (Gieskes et al., 1991) using a Milton Roy Spectronic 301 spectrophotometer and a Milton Roy Mr. Sipper sample introduction system. After phosphate values were found to be below detection limit for the first site, samples were no longer analyzed for phosphate. Manganese (Mn2+), lithium (Li+), strontium (Sr2+), barium (Ba2+), and boron (B) concentrations were determined by inductively coupled plasma-atomic emission spectroscopy (ICP-AES) following the general procedure outlined by Murray et al. (2000). In preparation for analysis by ICP-AES, aliquots of interstitial waters were acidified with nitric acid (HNO3) and diluted tenfold with deionized (DI) water (0.5 mL of sample and 4.5 mL of DI water). Analytical blanks were prepared identically by analyzing DI water, which was acidified to matrix match the samples. At all sites, sodium was determined using charge balance calculations, where
(Broecker and Peng, 1982). The chemical data for interstitial waters are reported in molar units. The reproducibility of results, determined via multiple determinations of the International Association for the Physical Sciences of the Ocean standard seawater (alkalinity, Cl-, Ca2+, Mg2+, K+, and SO42-), spiked synthetic seawater (ICP-AES determinations), or through the use of a calibration curve (NH4+, HPO42-, and Si[OH]4), is available in Table T7. Initially during Leg 199, reproducibility on interstitial water ICP-AES determinations (Mn2+, Li+, Sr2+, and Ba2+) was compromised because of analytical problems associated with the baffled spray chamber used for interstitial water ICP-AES. An unbaffled spray chamber more efficiently delivers the sample in the instrument, yields better reproducibility, and is normally used for interstitial water ICP-AES determinations. However, during Leg 199 the only available unbaffled spray chamber was sequestered for bulk-sediment ICP-AES determinations and could not be used for interstitial water analysis, because of concern over the possible contamination of the interstitial waters with lithium metaborate (LiBO2) flux from the bulk-sediment analyses. For these reasons, once the bulk-sediment analyses for Leg 199 were complete, we cleaned the unbaffled spray chamber with ~10% HNO3 and repeated the interstitial water program for Mn2+, Li+, Sr2+, Ba2+, and B for all samples. These repeat analyses are the ones that we report. The chemistry lab aboard the JOIDES Resolution now carries two unbaffled spray chambers to circumvent the above problems.
For the first time, bulk-sediment samples were analyzed routinely during Leg 199. Samples for bulk sediments were taken at a frequency of one sample per section (1.5 m) for Sites 1215-1217. To conserve shipboard argon (used in record quantity during Leg 199) samples were taken at a frequency of three samples per core (1.5 m; Sections 2, 4, and 6) for Sites 1218-1222. For future application of a shipboard bulk-sediment geochemistry program of this magnitude, we recommend significantly increasing the shipboard argon supply. All samples were taken adjacent to physical properties samples so that MARs can be calculated using concentration and bulk density measurements taken as close together as possible.
Bulk samples were prepared according to the method of Murray et al. (2000). Samples were first freeze dried and crushed. Samples and standards (0.1 g) were mixed with LiBO2 flux (0.4 g). Analytical blanks were prepared using 0.4 g LiBO2 flux to ensure matrix matching. A solution of 0.172-mM LiBr wetting agent (10 µL) was added to the samples, standards, and blanks. This mixture was fused for 15 min at 1050°C in a NT-2100 Bead Sampler prior to dissolution in 50 mL of 10% HNO3. The supply of platinum crucibles (12) for fusing was a significant limit on sample throughput. A 5-mL aliquot of the resulting solutions was filtered (0.45 µm) and diluted with 35 mL of 10% HNO3, resulting in a 4000x dilution of the original powders. Si, Al, Ti, Fe, Mn, Ca, Mg, P, Sr, and Ba concentrations were determined using a Jobin-Yvon Ultrace ICP-AES using a type C Meinhard concentric nebulizer following the general procedure outlined by Murray et al. (2000). We chose to report major element sediment concentrations in weight percent (Si, Al, Ti, Fe, Mn, Ca, Mg, and P) and minor element concentrations in parts per million (Sr and Ba) because sediments are composed of a mixture of minerals that are not usually in equilibrium. Reproducibility of results for Sites 1215-1221 is available in Table T7. We found at Site 1222 that excessive ship motion during transit or on station affected instrument drift and weighing accuracy enough to significantly degrade precision (e.g., from 11.5% to 34.2% for Ca, 14.7% to 85.4% for Sr, and 3.3% to 21.8% for Si). Therefore, we do not report results for Site 1222 and we recommend that during future legs these analyses not be run in heavy seas or during transit.
Sediment samples (at least one per core, on a split of one of the bulk-sediment samples) were analyzed for calcium carbonate (CaCO3 in weight percent) using a Coulometrics 5011 carbon dioxide coulometer and for total carbon using a Carlo Erba 1500 CNS analyzer. Organic carbon (in weight percent) was calculated as the difference between total carbon and carbonate carbon. CaCO3 was also calculated from Ca (in weight percent) determined by ICP-AES. The following equation (Dymond et al., 1976) was used:
where,
We compared CaCO3 values calculated from Ca determined by ICP-AES to CaCO3 values measured by coulometer (Fig. F9). CaCO3 values calculated from Ca contents yielded similar trends to CaCO3 measured via coulometer. However, absolute values by calculation are lower than by coulometer when CaCO3 is <1 wt% and higher when CaCO3 is >1 wt%. Values by calculation sometimes yield negative values because the normative calculation assumes an aluminosilicate calcium contribution of 0.7 wt%. Scatter is higher when CaCO3 values are >75 wt%. The discrepancy in higher values is probably due to a combination of factors, including the error on Ca measurements determined by ICP-AES (±11.5 wt%; see Table T7) and the fact that the highest Ca standard available for the ICP-AES calcium standard calibration curve was 35.9 wt%, equivalent to a value of 90 wt% CaCO3. Any reported calcium values >35.9 wt% should, therefore, be considered suspect and are flagged in this report. In the future, to alleviate this problem, we recommend using a 40 wt% Ca (100 wt% CaCO3) standard in the ICP-AES Ca standard calibration curve.
![]()