RESULTS AND DISCUSSION
Pore water 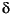 18O-SO4 values determined for Sites 1225, 1226, and 1231 are shown in Tables T1, T2, and T3 along with concentrations of dissolved sulfate, ammonium, sulfide, alkalinity, and metals. Methane was present at all of the sites, but only at low- to near trace-level amounts (<0.25 µM). The approach to normal seawater sulfate concentrations at depth is observed at all three of the sites, consistent with previous reports of basin-scale flow of modern seawater through basement and upward into the base of the sediment column (Baker et al., 1991; Oyun et al., 1995).
18O-SO4 values determined for Sites 1225, 1226, and 1231 are shown in Tables T1, T2, and T3 along with concentrations of dissolved sulfate, ammonium, sulfide, alkalinity, and metals. Methane was present at all of the sites, but only at low- to near trace-level amounts (<0.25 µM). The approach to normal seawater sulfate concentrations at depth is observed at all three of the sites, consistent with previous reports of basin-scale flow of modern seawater through basement and upward into the base of the sediment column (Baker et al., 1991; Oyun et al., 1995).
Site 1231 is the most organic carbon poor of all the Leg 201 sites and had the lowest measured microbial activity (D'Hondt, Jørgensen, Miller, et al., 2003; D'Hondt et al., 2004). As expected, Site 1231 also shows the smallest changes in concentration of dissolved constituents and 18O-SO4 values (Table T1; Fig. F2). Pore water sulfate concentrations decreased only slightly with depth, ranging from near bottom water values of ~29 mM over the uppermost 48-m interval, to ~27 mM over the lower 60-m interval of the core (Fig. F2). This slight decrease in sulfate concentration with depth may suggest some small amount of sulfate-reducing activity; however, sulfide (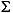 H2S = H2S(aq) + HS–) was below detection limits (0.0002 mM) in pore waters at Site 1231. 18O values of dissolved sulfate at Site 1231 remained essentially constant with depth (Fig. F2) and averaged about +10
H2S = H2S(aq) + HS–) was below detection limits (0.0002 mM) in pore waters at Site 1231. 18O values of dissolved sulfate at Site 1231 remained essentially constant with depth (Fig. F2) and averaged about +10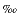 . These values are close to the currently accepted seawater value of +9.5 (Longinelli and Craig, 1967; Longinelli, 1989). Variations in alkalinity and ammonium concentrations with depth (Fig. F2) are attributed to chemical exchange processes (i.e., sinks) at both the sediment/seawater and sediment/basement interface (D'Hondt, Jørgensen, Miller, et al., 2003; D'Hondt et al., 2004). The isotopic composition of sulfur in dissolved sulfate (34S-SO4) at this site also reflects pore waters that are isotopically unmodified by MSR (Böttcher et al., this volume). Thus, several lines of chemical and stable isotopic evidence—sulfate and sulfide concentrations and 18O-SO4—clearly point to the absence of sulfate-reducing activity at open-ocean Site 1231.
. These values are close to the currently accepted seawater value of +9.5 (Longinelli and Craig, 1967; Longinelli, 1989). Variations in alkalinity and ammonium concentrations with depth (Fig. F2) are attributed to chemical exchange processes (i.e., sinks) at both the sediment/seawater and sediment/basement interface (D'Hondt, Jørgensen, Miller, et al., 2003; D'Hondt et al., 2004). The isotopic composition of sulfur in dissolved sulfate (34S-SO4) at this site also reflects pore waters that are isotopically unmodified by MSR (Böttcher et al., this volume). Thus, several lines of chemical and stable isotopic evidence—sulfate and sulfide concentrations and 18O-SO4—clearly point to the absence of sulfate-reducing activity at open-ocean Site 1231.
Equatorial Upwelling Site 1225
There is chemical and isotopic evidence for increased microbial activity at Site 1225 when compared to Site 1231 that is consistent with the abundance of higher amounts of organic matter found at this higher-bioproductivity site (D'Hondt, Jørgensen, Miller, et al., 2003; D'Hondt et al., 2004). Pore water sulfate concentrations decrease with depth from ~29 mM at 7.3 mbsf to a minimum of 27 mM at 159.3 mbsf, then rise again to 28 mM at the sediment/basement interface near 319 mbsf (Fig. F3). 18O-SO4 values increase gradually with depth from +9.5 at 1.5 mbsf to a plateau of +20 between ~178 and 250 mbsf, indicating significant activity of sulfate-reducing bacteria (Table T2; Fig. F3). Below this plateau, 18O-SO4 values decrease more rapidly with depth and approach a normal seawater value (+9.9) at ~319 mbsf. The sulfate concentration profile mirrors the 18O-SO4 vs. depth profile in its overall shape, and the maximum 18O-SO4 plateau coincides with the zone of minimum sulfate concentration (~27 mM). The return of 18O-SO4 values and pore water sulfate concentrations to normal seawater values at the base of the sediment column further supports previous findings that indicate the flow of modern seawater through underlying basaltic basement rocks in the eastern equatorial region.
Although high 18O-SO4 values (as high as +20) are a strong indication of MSR at Site 1225, pore water sulfate concentration and alkalinity vs. depth profiles at this site are nearly identical to those at the low-activity open-ocean Site 1231, which had typical seawater 18O-SO4 values and showed no evidence of MSR.
The process of dissimilatory MSR should result in increased concentrations of sulfide, bicarbonate (i.e., alkalinity; Equation 1), and products of organic matter degradation such as ammonium.
-
2CH2O + SO42–
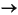 2HCO3– + H2S. (1)
2HCO3– + H2S. (1)
Alkalinity increased very little with depth from normal seawater values (~3–4 mM); however, ammonium concentrations increased from 6 µM at 1.5 mbsf to a maximum of 76 µM at 159 mbsf, which is twice the maximum value observed at Site 1231 (Table T2; Fig. F2). One explanation for the difference in 18O-SO4 behavior at Sites 1225 and 1231, despite similar sulfate and alkalinity profiles, is that sulfide produced from MSR at Site 1225 has been oxidized completely back to sulfate without diffusional loss of sulfide. This would result in diminished net reduction in the concentration of pore water sulfate (Ku et al., 1999). Dissolved sulfide was below detection at Site 1225, which supports its removal by iron sulfide formation, sulfide oxidation, and/or some alternative process occurring within the sediments at this site. However, sulfur isotope discrimination in dissolved residual sulfate is consistent with minor net MSR taking place in the pore waters at Site 1225 (Böttcher et al., this volume).
The oxygen in sulfate produced by oxidation of sulfide may be acquired from dissolved oxygen, water, or a mixture of the two, depending on the oxidation pathway (biotic vs. abiotic) and superimposed by contributions from intermediate processes of sulfur disproportionation, which in turn may depend on the involvement of reactive mineral oxidants (e.g., Fe or Mn oxides) present in the sediments (Taylor et al., 1984; van Stempvoort and Krouse, 1994; Böttcher et al., 2001, 2004; Böttcher and Thamdrup, 2001). The 18O values of sulfate derived from sulfide oxidation depend further on the fractionation of oxygen isotopes between sulfate and the oxidant source (i.e., water or O2) (Taylor et al., 1984; van Stempvoort and Krouse, 1994). Controlled experiments on the oxidation of various sulfur compounds (e.g., elemental sulfur, sulfite, and pyrite) indicate that water is the dominant source of oxygen in sulfate derived from oxidation of sulfide and elemental sulfur (van Stempvoort and Krouse, 1994; Böttcher et al., 2001, 2004; M. Böttcher and A. Schippers, unpubl. data).
Dissolved sulfate 18O values at Site 1225 seem to reflect a mixture of sulfate contributed from several different sources/processes, including sulfate diffusing upward from the sediment/basement interface and downward from the sediment/water interface, residual sulfate remaining after MSR, sulfate derived from sulfide oxidation, and sulfate that has undergone enzyme-catalyzed exchange with ambient pore water. Sulfate diffusing into the sediment column from either above or below is derived from normal seawater and, thus, has a 18O value of about +9.5 (Fig. F3). The 18O value of residual sulfate following MSR is determined by the fractionation during MSR. Apparent fractionation factors, 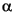 , for oxygen isotopes in sulfate have been determined for pure cultures of sulfate-reducing bacteria and from enrichments of marine sediments (e.g., Kemp and Thode, 1968; Mizutani and Rafter, 1973; Ku et al., 1999; M.E. Böttcher and J. Detmers, unpubl. data, 2001; M.E. Böttcher, J. Benecke, and H. Cypionka, unpubl. data). The oxygen isotope fractionation factor is estimated in the present study assuming a closed system and using a simplified Rayleigh equation (Rayleigh, 1896; Mizutani and Rafter, 1973; Goldhaber and Kaplan, 1974; Aharon and Fu, 2000)
, for oxygen isotopes in sulfate have been determined for pure cultures of sulfate-reducing bacteria and from enrichments of marine sediments (e.g., Kemp and Thode, 1968; Mizutani and Rafter, 1973; Ku et al., 1999; M.E. Böttcher and J. Detmers, unpubl. data, 2001; M.E. Böttcher, J. Benecke, and H. Cypionka, unpubl. data). The oxygen isotope fractionation factor is estimated in the present study assuming a closed system and using a simplified Rayleigh equation (Rayleigh, 1896; Mizutani and Rafter, 1973; Goldhaber and Kaplan, 1974; Aharon and Fu, 2000)
-
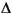 = t – 0 = 103( – 1) ln(f), (2)
= t – 0 = 103( – 1) ln(f), (2)
where
t = 18O value of residual sulfate at time t,
0 = initial 18O value of sulfate before any MSR, and
f = fraction of sulfate remaining at time t (f = 1 at t = 0).
The apparent fractionation is obtained from the slope of this equation, 103( – 1), which is expressed in terms of the isotope enrichment factor, 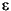 (in permil).
(in permil).
Data from the upper 170-m zone of Site 1225, plotted according to Equation 2, show nonlinear behavior that suggests variable fractionations and that more than simple MSR is affecting 18O-SO4 values at this site (Fig. F4A). The -SO4 determined for this zone is +54 (Fig. F4B), which is significantly larger than previously reported -SO4 for MSR of +4 to +29 (e.g., Lloyd, 1967; Fritz et al., 1989). The multiple and potentially complex pathways for sulfide oxidation (e.g., Böttcher et al., 2001) make it difficult to constrain -SO4. Accordingly, very few values for -SO4 have been reported in the literature (Ku et al., 1999; Aharon and Fu, 2000), and to date none have been determined for sediment depths >50 mbsf. The total range of reported -SO4 is –8.7 to +29 and includes biotic and abiotic pathways for oxidation of sulfide, sulfite, and elemental sulfur, using both water and dissolved oxygen as the oxygen source (van Stempvoort and Krouse, 1994; Ku et al., 1999; Aharon and Fu, 2000; Böttcher et al., 2001, 2004; Böttcher and Thamdrup, 2001). The -SO4 determined for Site 1225 (+54) is higher than previously reported values by almost more than a factor of two. This very steep slope (+54) is accompanied by a small amount (~2 mM) of net sulfate removal (i.e., high f [sulfate] values) and further supports the occurrence of sulfide oxidation at this site as an explanation for both the heavy sulfate 18O values and the relative lack of change in sulfate concentration. The present results make clear that no constant apparent fractionation factor exists and that specific sets of biogeochemical conditions may be superimposed to produce different relations between the oxygen isotope composition of dissolved sulfate and net sulfate reduction.
Several potential oxidants for sulfide are present in both the upper and lower regions of the sediment column at Site 1225 and include iron, manganese, nitrate, and dissolved oxygen. These oxidants may form an upper and lower oxidizing boundary for sulfide diffusing away from the MSR zone. For example, there is a broad peak in the concentration of dissolved iron (Fe2+) between 175 and 275 mbsf, which coincides with the zone of maximum 18O-SO4 and, presumably, maximum MSR, and a less pronounced peak centered at ~20 mbsf (Fig. F3). Sulfide produced from MSR may have reduced sedimentary reactive Fe3+ oxide phases to Fe2+, which subsequently removed additional sulfide (not detected at Site 1225) as iron-sulfide phases. There is also a large peak in dissolved Mn between 1.5 and 64.3 mbsf and a smaller peak below the MSR zone between ~237 and 278 mbsf that could have served as a reaction sink for sulfide at this site (Fig. F3).
A quantitative evaluation of the results is complicated by the fact that sulfate reduction in the deep sediments takes place under partly open conditions (Böttcher et al., this volume), with sulfate diffusing from the top and the base into the sediment section. The downcore trend in oxygen isotope values is also consistent with an enzymatically catalyzed isotope exchange upon bacterial sulfate reduction, finally leading to an isotopic equilibration. The observed enrichment of the heavy oxygen isotope, however, is of unusual magnitude when compared to the small net amount of sulfate reduced. This may be explained by a superimposition of MSR by disproportionation processes that are associated with an enrichment of 18O in the secondary sulfate formed (Böttcher et al., 2001). A second explanation has been introduced by Böttcher et al. (1998a), who suggested that the relative degree of oxygen isotope equilibration may be controlled by microbial sulfate reduction rates. Therefore, at lower cellular rates, more time is provided for intracellular oxygen isotope exchange. Maximum oxygen isotope values of sulfate are still below the exchange equilibrium values (Fritz et al., 1989; Böttcher et al., 1998a) expected at maximum temperatures of 7°C observed at Site 1225 (D'Hondt, Jørgensen, Miller, et al., 2003). This indicates that the low MSRs, mixing of sulfate from different extents of equilibration, and/or sulfide oxidation did not lead to complete stable isotope exchange equilibrium. However, the observed oxygen isotope data make it possible to clearly distinguish the activity of the biosphere in the deep sulfur cycles of Sites 1225 and 1231.
Equatorial Upwelling Site 1226
Site 1226 is the most organic rich of the eastern equatorial Pacific sites and is also located at the shallowest water depth (3297 m). Pore water 18O-SO4 and sulfate concentration profiles are clearly more influenced by sulfate reduction at this site (Table T3; Fig. F5). 18O-SO4 values increase with depth from +11.8 at 1.3 mbsf to a plateau of +28 between ~70 and 140 mbsf, and then decline toward the bottom of the hole due to introduction of seawater that flows through the underlying basaltic basement without obvious modification by bacterial sulfate reduction. 18O-SO4 values reach +12.7 at the maximum depth drilled of 418 mbsf (Fig. F5). Higher maximum 18O-SO4 values at Site 1226 compared with Site 1225 are consistent with greater extents of MSR at Site 1226 facilitated by a higher amount of sedimentary organic matter. Dissolved sulfate concentrations generally mirror the 18O-SO4 profile, declining from 30.1 mM at 1.3 mbsf to a minimum of 19.8 mM at 246.2 mbsf and then rising with depth to reach 25.2 mM at 418 mbsf (Fig. F5). Both increased alkalinity and significant amounts of dissolved sulfide were detected in the uppermost ~250 m at Site 1226 (Fig. F5). Dissolved sulfide formed a broad maximum peak of 600–700 µM between 45 and 130 mbsf (Fig. F5). A plot of dissolved sulfate vs. sulfide (Fig. F6) shows a negative linear correlation, which suggests that sulfide is forming at the expense of sulfate; however, the slope of this line is an order of magnitude smaller than the slope of one expected for a MSR-dominated system. Data points are concentrated far below a 1:1 slope line, indicating significant loss of sulfide, most likely due to formation of iron sulfides, and probably a partial diffusion out of the maximum MSR zone and subsequent oxidation at shallower depths. The isotopic and chemical profiles at Site 1226 indicate, similar to Site 1225, that dissolved sulfate enters the sediment column from both above (sediment/water interface) and below 418 mbsf (sediment/basement interface) and is reduced by microbial activity within the sediments.
Sulfate oxygen isotope enrichment factors (-SO4) at Site 1226 are +42.0 in the upper MSR zone and +79.4 in the lower zone (below ~200 mbsf) (Fig. F7). Similar to Site 1225, these -SO4 values are higher than the previously reported range of values, –8.7 to +29, and likely reflect bacterial sulfate reduction at low cellular sulfate reduction rates, probably superimposed by sulfide oxidation and disproportionation reactions of sulfur intermediates. Assuming normal seawater 18O values of 0 for the pore waters, the theoretical equilibrium value between sulfate and water is still not reached at Site 1226 but is more closely approximated than at Site 1225. Besides the higher extent of net sulfate reduction observed at this site, the enhanced temperatures may have additionally influenced the overall shape of the isotope profiles. The higher temperatures may enhance microbial sulfate reduction rates (J. Kallmeyer and T.G. Ferdelman, unpubl. data, 2004), and in addition a lower 18O-SO4 value is expected under equilibrium conditions (e.g., Fritz et al., 1989). A complete quantitative evaluation of the observed stable oxygen isotope fractionation, however, requires additional consideration of system openness and associated differential transport processes in the pore waters, as already shown for the stable isotopes of sulfur (Chanton et al., 1987; Jørgensen, 1979; Jørgensen et al., 2004) and the combination of oxygen isotope measurements with results for sulfur isotope compositions (Böttcher et al., this volume).
