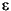 ,
, 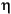 , and
, and 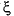 ; see Fig. F1) appear to consist of sediment and subsurface phylotypes, suggesting evolutionary diversification of these MG-I subgroups in marine sediments and subsurface environments (Sørensen et al., 2004; A. Lauer, unpubl. data).
; see Fig. F1) appear to consist of sediment and subsurface phylotypes, suggesting evolutionary diversification of these MG-I subgroups in marine sediments and subsurface environments (Sørensen et al., 2004; A. Lauer, unpubl. data).
Because of its high degree of conservation, high information content, and good agreement with most physiological and genetic markers, the 16S ribosomal ribonucleic acid (rRNA) gene is the most widely used molecular marker to infer phylogenetic relationships in the living world (Woese, 1987) and provides the basis for the three-domain tree of life, with bacteria, archaea, and eukaryotes as the largest phylogenetic units (domains) of life (Woese et al., 1990). The current census of the microbial diversity of life, based on 16S rRNA genes of pure cultures and natural mixed populations in environmental samples, includes at least 52 phylum-level bacterial and ~20 phylum-level archaeal phylogenetic lineages, most of them environmental populations not available in pure culture (Rappe and Giovannoni, 2003; Hugenholtz et al., 1998; Hugenholtz, 2002). In other words, only a small portion of the microbial world has been brought into pure culture and studied biochemically or physiologically. These phylum-level lineages go back to the deep, early radiations of the bacterial and archaeal domains where the phylogenetic resolution of the 16S rRNA molecule breaks down, in the sense that a hierarchical branching pattern cannot be obtained. The number of these mutually exclusive phylogenetic lineages may increase in the near future, as a function of more comprehensive sequencing surveys.
For microbial community analyses of deep subsurface environments, deoxyribonucleic acid (DNA) recovery is usually the critical factor. DNA has to be extracted from deep subsurface sediments, usually by enzymatic, mechanical, or freeze-thawing lysis of cells, followed by removal of lipids and proteins by extraction with organic solvents (phenol and chloroform) and precipitation of DNA in the aqueous phase with salts and alcohols at cold temperatures. Because of low DNA content and recovery, standard methods must be fine-tuned empirically for optimized DNA recovery from deep subsurface samples (for detailed discussion of empirically optimized protocols, see Sørensen et al., 2004, and Webster et al., 2003). DNA extraction, amplification of 16S rRNA genes by polymerase chain reaction (PCR), cloning, and sequencing of selected clones yield individual 16S rRNA gene sequences of uncultured bacteria and archaea that occur in a specific sediment sample (Sørensen et al., 2004; Newberry et al., 2004; Webster et al., 2003). Sequence alignments and phylogenetic analyses of the 16S rRNA gene sequences are performed with several software packages as detailed in the original references of Tables T1 and T2, which summarize the results in terms of percent representation of archaeal and bacterial lineages in clone libraries. A necessary caveat is that because of potential biases in nucleic acid extraction, PCR amplification, and cloning efficiency, the percent representation of a bacterial or archaeal phylogenetic lineage in a clone library cannot be equated with the relative abundance of these cells in the environment. So far, clone libraries are used as a first approximation to describe the microbial community; group-specific fluorescent in situ hybridization (FISH) counts give direct, quantitative information on community composition (Amann et al., 1995; Pernthaler and Amann, 2005).
16S rDNA clone libraries from all Leg 201 sites were dominated by diverse uncultured lineages of bacteria and archaea. Several phylum-level archaeal lineages recur consistently in deep subsurface environments and Leg 201 subsurface sediments. The phylogenetic tree (Fig. F1) shows these archaeal lineages, illustrated with selected sequences from Sites 1225 and 1231. Table T1 lists the percent representation of archaeal subsurface lineages in 16S rDNA clone libraries from Leg 201 sediment samples. For comparison, clone library data from other marine subsurface sediments are included.
Members of the Deep-Sea Archaeal Group (DSAG) are conspicuously well represented in clone libraries of archaeal 16S rRNA genes from diverse sampling sites and sediment types. DSAG archaea were originally found at hydrothermal vent sites (Takai and Horikoshi, 1999) and appear in a growing number of molecular surveys of deep subsurface and hydrothermal vent sites. In addition to the examples shown in Table T1, the deep-sea archaeal group represents >50% of all archaeal clones in 16S rDNA clone libraries at ODP Leg 204 Sites 1245 and 1251 on Hydrate Ridge (Inagaki et al., 2006). Further, DSAG archaeal clones were the second largest archaeal group (13%) recovered in clone libraries from surficial Atlantic deep-sea sediments (Vetriani et al., 1999). Thus, DSAG archaea show a conspicuously cosmopolitan occurrence pattern in a wide spectrum of sediments and vents. For Leg 201, DSAG archaea occur from organic-poor sediments of the central oceanic basins (Sites 1225 and 1231) (Fig. F1) to predominantly organic-rich, methane- or methane hydrate–containing sediments near continental margins (Sites 1230, 1245, and 1251). As an exception to this pattern, Peru Margin Sites 1227 and 1229 appear to be dominated by archaeal groups other than DSAG, specifically members of the Miscellaneous Crenarchaeotal Group (MCG) and of the South African Goldmine Euryarchaeotal Group (SAGMEG) archaea (Inagaki et al., 2006; Parkes et al., 2005; Sørensen and Teske, in press).
The second major archaeal lineage that is frequently found in subsurface sediments are the Marine Group I (MG-I) archaea (Table T1). Members of this group were originally identified by sequencing of environmental rRNA from seawater (DeLong, 1992; Fuhrman et al., 1992). MG-I archaea account for a major portion of all prokaryotic picoplankton in seawater (DeLong et al., 1994; Fuhrman and Ouverney 1998; Karner et al., 2001). In the deep-sea water column below ~3000 m depth, MG-I archaea constitute the majority of prokaryotic picoplankton (Karner et al., 2001). Although pure culture studies are not available, members of MG-I have been shown to take up amino acids, indicating potential for heterotrophic nutrition (Ouverney and Fuhrman, 2000). However, stable C isotope analyses of MG-I lipids suggested that these archaea utilize inorganic carbon (Pearson et al., 2001), which matches the recent finding that Marine Group archaea are capable of HCO3– uptake, suggesting autotrophic CO2 fixation (Wuchter et al., 2003). Thus, MG-I archaea might be facultative autotrophs, or there is broad metabolic diversity within this group. Recently, the first isolate of this group was described as an aerobic ammonia oxidizer with the capability for autotrophic growth (Könneke et al., 2005). Since MG-I archaea are abundant in seawater, retrieving their 16S rRNA genes from subsurface clone libraries poses the question whether (1) they could be seawater contaminants that are introduced into the sediments during the drilling process; (2) seawater archaea that permeate the subsurface naturally, for example by entrainment in subsurface flow through basement basalt, and tolerate the subsurface conditions well enough to persist in this environment; or (3) native archaea of the subsurface that originate and grow in this environment. The second and third possibility are not unreasonable, since the number of 16S rRNA phylotypes of MG-I archaea in subsurface samples (Sites 1225 and 1231) exceeds the number of prokaryotic cells that could have contaminated the samples based on perfluorocarbon tracer (PFT) assays (Sørensen et al., 2004). Also, some phylogenetic clusters within the MG-I archaea (clusters , , and ; see Fig. F1) appear to consist of sediment and subsurface phylotypes, suggesting evolutionary diversification of these MG-I subgroups in marine sediments and subsurface environments (Sørensen et al., 2004; A. Lauer, unpubl. data).
In contrast to DSAG and MG-I, archaea of the Marine Benthic Groups A (MBG-A) and D (MBG-D) have been detected in fewer samples and sites (Table T1) and usually do not dominate deep subsurface clone libraries. Originally, they were found in 16S rDNA surveys of push cores retrieved from surficial sediments (upper 30 cm) of the Atlantic continental slope and abyssal plain offshore New England (Vetriani et al., 1999). Clones of these groups occur in deep subsurface sediments of Leg 201 (MBG-A at Site 1225, see Figure F1; MBG-D at Sites 1227 and 1230). In contrast to MG-I archaea, they are not detected in the water column and they appear to be benthic, sediment-dwelling archaea. Interestingly, a clone of MBG-A was found in enrichment cultures inoculated with Site 1230 sediment and incubated under aerobic conditions at 10°C (Biddle et al., this volume).
Several uncultured lineages appear in molecular studies of marine, as well as terrestrial, deep subsurface environments. For example, members of the MCG and SAGMEG lineages (Fig. F1) have been found in Leg 201 sediments, especially at Sites 1227 (Inagaki et al., 2006; Sørensen and Teske, in press) and 1229 (Parkes et al., 2005), but also in Mediterranean sapropel sediments (Coolen et al., 2002), and in the deep terrestrial subsurface, such as South African Goldmines (Takai et al., 2001). A similarly mixed habitat range applies to archaea of the Terrestrial Miscellaneous Euryarchaeotal Group (TMEG) lineage (Fig. F1), which have been found in a wide range of terrestrial and freshwater environments and marine subsurface sediments, including Site 1231 (Takai et al., 2001; Sørensen et al., 2004). None of these archaea are cultured, and their physiology remains unknown. It is hoped that the designation "Miscellaneous" in the original names of these archaeal lineages (Takai et al., 2001) will change to a more informative label that reflects the very wide environmental occurrence of these interesting but elusive generalists.
In a few cases, archaeal phylotypes from Leg 201 sediments are specifically related to archaea from hydrothermal vents. Examples include Methanocaldococcus-related 16S rDNA clones from Site 1230 (Inagaki et al., 2006) and archaeal phylotypes from Site 1231 (members of the "Peru Basin cluster") (Fig. F1) that form a monophyletic lineage with an archaeal clone from hydrothermal vent fluids at Juan de Fuca (Huber et al., 2002). These observations pose interesting questions about links between nonhydrothermal subsurface sediments and hydrothermal habitats. As general working hypotheses, hydrothermal archaea may reach deep nonhydrothermal subsurface sediments by subsurface fluid flow, potentially through conduits in basement basalt, following the same flow paths as chemical oxidants (D'Hondt et al., 2004). Conversely, nonhydrothermal sediment archaea could become entrained in hydrothermal circulation and are then identified in vent fluids (Sørensen et al., 2004).
Among the bacteria, the recently identified candidate division JS-1 (Webster et al., 2004) and the Chloroflexi division (divided into four subphyla) (Hugenholtz et al., 1998; Rappe and Giovannoni, 2003) are well represented in 16S rDNA clone libraries at Leg 201 sites and other subsurface locations (Table T2). The only cultured members of the Chloroflexi Subphyla I, II, and IV include the anaerobic, H2-dependent dehalogenating bacterium Dehalococcoides ethenogenes (Maymó-Gatell et al., 1997) and thermophilic filamentous bacteria that grow chemoheterotrophically on diverse carbohydrates (Sekiguchi et al., 2001, 2003). No cultured member of the JS-1 group is known at present. Of all bacterial phyla found in the deep subsurface, only proteobacterial subsurface clones are sometimes closely related to cultured species, allowing physiological inferences.
The JS-1 division and the Chloroflexi division are consistently found in diverse subsurface environments, using different methodologies. In addition to the examples in Table T2, members of the Chloroflexi accounted for as much as 69% of total DNA detected by quantitative PCR testing in subsurface sapropel layers of the eastern Mediterranean (Coolen et al., 2002). The JS-1 candidate division was detected with group-specific primers in marine sediments worldwide; deep subsurface sediments as well as in coastal surficial sediments (Webster et al., 2004, and references therein). The uncultured members of the Chloroflexi and JS-1 candidate divisions also predominate at the Peru margin sites.
The conspicuous methane/sulfate gradients in Leg 201 subsurface sediments have led to working hypotheses that guided initial molecular community surveys. In brief, these large-scale subsurface gradients were thought to be dominated by sulfate-reducing, methanogenic, and methane-oxidizing microbial communities with a clear stratification. Sulfate-reducing prokaryotes should dominate in the sulfate-containing upper sediment layers (Bale et al., 1997; Barnes et al., 1998); methanogenic archaea were expected in the methane-enriched deeper sediment layers, as in some previous subsurface surveys (Marchesi et al., 2001); and sulfate-dependent, methanotrophic consortia analogous to those found at methane seeps and vents (Hinrichs et al., 1999; Boetius et al., 2000; Orphan et al., 2001, 2002; Teske et al., 2002; Michaelis et al., 2002) were expected to dominate the sulfate–methane transition zones. Consequently, 16S rDNA surveys were complemented by functional key gene surveys targeting key genes of sulfate reduction and methanogenesis, dissimilatory sulfate reductase (dsrAB) (Wagner et al., 1998; Klein et al., 2001; Zverlov et al., 2005) and methyl-coenzyme M reductase (mcrA) (Springer et al., 1995; Hales et al., 1996; Luton et al., 2002). Partially modified mcrA primers also cover the mcrA genes of anaerobic, sulfate-dependent, methane-oxidizing archaea (Hallam et al., 2003).
Interestingly, 16S rRNA genes of sulfate-reducing prokaryotes and methanogenic archaea were found only in relatively few cases and suggested that these populations constitute only a minor component in subsurface microbial ecosystems dominated by novel, uncultured archaeal and bacterial phylum-level lineages. Identifiable methanogens included Methanocaldococcus-related phylotypes in the deepest sediment layers (below 200 mbsf) at Site 1230 (Inagaki et al., 2006). Evidence for sulfate-reducing bacteria was similarly spotty. Small numbers of delta-Proteobacterial clones that potentially represent sulfate reducers were found by 16S rRNA gene amplification at Sites 1227 and 1230 but with no apparent depth stratification that mirrored sulfate gradients. At Site 1230, deep, sulfate-free sediment layers (below 150 mbsf) yielded more delta-Proteobacterial clones than the surface layers (Inagaki et al., 2006).
The detection of methanogens and sulfate reducers required the use of primers for key genes of sulfate-reducing and methanogenic pathways. The key gene of methanogenesis, mcrA, was detected with specific primers in nested PCR assays at a few depth horizons at Sites 1229 and 1230. The mcrA phylotypes were related to members of the genera Methanobrevibacter and Methanosarcina at Site 1229 (Parkes et al., 2005) or formed a sister group to the genus Methanosaeta at Site 1230 (Lever and Teske, 2005; Inagaki et al., 2006). As at other ODP sites, the detection of methanogens required the use of group-specific or selective PCR primers, either for methanogen 16S rRNA genes (Marchesi et al., 2001) or for mcrA genes (Newberry et al., 2004). Interestingly, the mcrA phylotypes at sites from ODP Leg 190 (Newberry et al., 2004) were most closely related to Methanosarcina and Methanobrevibacter genera, as at Site 1229 (Parkes et al., 2005). Methanosarcina species use acetate, methylated compounds, and H2/CO2 as substrates of methanogenesis (Boone and Mah, 2001); Methanosaeta species grow strictly by acetoclastic methanogensis (Patel, 2001); and Methanobrevibacter species use H2 and occasionally formate as methanogenic substrates (Miller, 2001).
Given the dominance of novel, uncultured bacterial and archaeal 16S rRNA gene lineages in all clone libraries recovered from Leg 201 samples, the working hypothesis that deep subsurface sediments are dominated by classical sulfate-reducing, methanogenic, and methane-oxidizing communities appears to be problematic. The most parsimonious explanation is that these classic anaerobic communities exist in low population densities and activities in deep subsurface sediments and create the conspicuous methane–sulfate gradients that have guided the sampling schemes for Leg 201. Very low rates of methanogenesis and sulfate reduction are sufficient to maintain these sulfate and methane profiles (D'Hondt et al., 2004). The dominant populations of novel, uncultured bacteria and archaea in the Leg 201 sediments may not be involved in these processes at all and could rely on other metabolic processes, such as fermentation (Biddle et al., 2006). Alternatively, at least some of the uncultured bacterial and archaeal lineages in Leg 201 sediments could be sulfate reducers, methanogens, or methane oxidizers, perhaps with highly altered key genes that escape PCR detection.
![]()