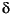 37Cl and 129I) as well as hydrate formation and dissociation (18O and D/H) processes in this environment.
37Cl and 129I) as well as hydrate formation and dissociation (18O and D/H) processes in this environment.
The biogeochemistry program of Leg 204 had three major objectives: (1) to explore the relationship between fluid flow and gas hydrate dynamics at Hydrate Ridge, (2) to constrain the role of microbial communities in methane generation and consumption, and (3) to use carbonate and sulfide minerals as potential indicators of past events of hydrate formation and dissociation. The distribution of dissolved constituents in IW has proven to be a powerful tool for quantifying hydrogeochemical and geological processes within sediments. The sampling, processing, and preservation protocol for IW samples during this leg were designed to provide the data needed to address these biogeochemical objectives.
Dissolved chloride content of the pore fluids has been widely used to constrain estimates of hydrate concentration in marine sediments (e.g., Froelich et al., 1995; Kastner et al., 1995; Paull, Matsumoto, Wallace, et al., 1996; Hesse, 2003). These estimates are based on the concept that dissolved ions are excluded from the hydrate cages during gas hydrate formation, such that the gas hydrate lattice contains little or no salt. This process causes the pore water to become saltier, and over time, the excluded ions are removed from the zones of hydrate formation by advection or diffusion. Gas hydrate dissociation during core recovery results in freshening of the pore fluids by the addition of water sequestered by gas hydrate prior to recovery (Hesse and Harrison, 1981). Thus, negative chloride anomalies relative to in situ chloride concentration should be proportional to the amount of gas hydrate in a sediment sample. During Leg 204, high-resolution profiles of dissolved chloride were used to generate first-order estimates of the amount of hydrate in sediment. Uncertainties in these estimates arise from a paucity of information on (1) the in situ chlorinity values of pore water; (2) the amount of chloride potentially trapped within the gas hydrate internal structure, such as micropores or veins; (3) the spatial sampling bias; and (4) fluid transport and diffusion rates. Shore-based isotopic characterization of the pore fluids will provide additional constraints on physical transport (e.g., 37Cl and 129I) as well as hydrate formation and dissociation (18O and D/H) processes in this environment.
Methane hydrate formation depends on pressure, temperature, pore water salinity, and a sufficient amount of methane. Methane inventory is highly dependent on rates of microbial methanogenesis, microbial methane oxidation, and physical transport processes. The dissolved sulfate distribution in marine pore fluids can be used to estimate methane flux toward the seafloor in certain situations, which, in turn, depends on the methane inventory below (Borowski et al., 1996). The distribution of dissolved sulfate in Leg 204 IW samples has been used to provide constraints on methane fluxes on Hydrate Ridge. Shore-based isotopic characterization of dissolved carbon, nitrogen, and sulfur species as well as data on microbial communities and metabolic rates will further constrain preliminary shipboard estimates.
The majority of shipboard IW samples were obtained on 5- to 20-cm-long whole-round cores that were cut according to two general procedures. Routine samples at each site were collected at a frequency of approximately two sections per core in the upper 150 m of the sediment section, followed by a sampling resolution of one whole-round sample per core in the deeper sequences. These whole-round samples were cut on the catwalk, capped, and taken to the laboratory for immediate processing. At most sites, samples within the anaerobic methane oxidation (AMO) zone were taken at higher resolution (approximately two samples per section) in a coordinated program with the shipboard microbiologists. Samples from the end of each section were cut on the catwalk, and the samples from the middle intervals of each section were cut from microbiology whole-round sections that were subsampled in the walk-in refrigerator. During high-resolution sampling, when there were too many IW cores to process immediately, capped whole-round core sections were stored in a refrigerator until they were squeezed, which occurred no later than 12 hr after core retrieval.
After extrusion from the core liner, the surface of each whole-round IW core sample was carefully scraped with an acid-washed (10% HCl) plastic spatula to remove potential contamination from seawater and sediment smearing in the borehole. In APC cores, ~0.5 cm from the outer diameter, top and bottom faces were removed; whereas in the XCB cores, where borehole contamination is higher, as much as two-thirds of the sediment was removed from each whole round. Approximately 2 cm3 of uncontaminated sediment was saved for grain-size analyses. The remaining sediment (~150-300 cm3) was placed into a titanium squeezer, modified after the stainless-steel squeezer of Manheim and Sayles (1974). Gauge pressures up to 15 MPa were applied using a laboratory hydraulic press to extract pore water. IW was passed through a prewashed Whatman No. 1 filter fitted above a titanium screen, filtered through a 0.45-µm Gelman polysulfone disposable filter, and subsequently extruded into a precleaned (10% HCl) 50-mL plastic syringe attached to the bottom of the squeezer assembly. In most cases, 25-40 cm3 of pore water was collected from each sample, which required squeezing the sediment for 20-40 min. To prevent loss of ephemeral constituents, IW subsamples were collected in a 10-mL syringe attached to the squeezer assembly and the 50-mL syringe via a three-way valve (D'Hondt, Jørgensen, Miller, et al., 2003). IW emerging from the disposable filter at the bottom of the squeezer assembly was directed into one or the other syringes, as necessary, for rapid and appropriate dispensing of aliquots. Use of the 10-mL syringe also avoided air bubbles, minimized contamination of this fraction of the IW by dissolved oxygen, and allowed for efficient subsampling during the sediment squeezing process (D'Hondt, Jørgensen, Miller, et al., 2003).
In addition to the whole-round IW samples, we collected 10-cm3 samples from the working half of the cores within 90 min after core retrieval at Sites 1245 and 1249. To construct a high-resolution chloride distribution relative to a discrete gas hydrate layer, we collected samples from the hydrate and at various distances from it in Core 204-1245C-7H. To further evaluate the effect of heterogeneous gas hydrate distribution and to recover sediments that showed minimum disturbance from gas hydrate dissociation, we collected paired samples from consolidated and mousselike sediments from Cores 204-1249F-3H and 7H. These samples were squeezed following the same procedures as described for the whole-round samples, but the amount of fluid collected (<5 cm3) was only enough for analyses of dissolved chloride (shipboard) and characterization of the isotopic composition of the water (shore based).
Subsamples were collected from the whole-round IW samples for shore-based isotopic characterization of the water (oxygen and deuterium) and dissolved metabolites (bicarbonate, sulfate, sulfide, and ammonium). These subsamples were preserved with mercuric chloride or cadmium acetate and were flame sealed in glass ampoules. In addition, subsamples were collected for analyses of dissolved volatile fatty acids (in glass vials and frozen), halogens (including 37Cl and 129I), and minor and trace metal constituents (acidified with ultrapure nitric acid). Dissolved sulfide (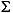 HS-) within pore water was fixed by pipetting a 1-mL IW subsample into a vial containing 1.0 mL of 1-M cadmium acetate solution (20 g ZnAc per 100-mL solution) and will be measured onshore using the methylene blue method of Cline (1969).
HS-) within pore water was fixed by pipetting a 1-mL IW subsample into a vial containing 1.0 mL of 1-M cadmium acetate solution (20 g ZnAc per 100-mL solution) and will be measured onshore using the methylene blue method of Cline (1969).
IW samples were analyzed for routine shipboard measurements according to standard procedures (Gieskes et al., 1991). Salinity was measured as total dissolved solids using a Goldberg optical handheld refractometer. The pH was determined by ion-selective electrode. Alkalinity was determined by Gran titration with a Metrohm autotitrator. Sulfate (SO42-) concentration was measured using a Dionex DX-120 ion chromatograph. High-precision chloride concentrations were determined by titration using silver nitrate (AgNO3), and the values were corrected for the presence of the other halogens assuming seawater ratios, as detailed by Gieskes et al. (1991). International Association of Physical Sciences organization (IAPSO) standard seawater was used for calibrating sulfate and chloride techniques. Dissolved phosphate (PO43-) and ammonium (NH4+) concentration were determined by spectrophotometric methods using a Milton Roy Spectronic 301 spectrophotometer, with a "Mr. Sipper" sample-introduction system (Gieskes et al., 1991). During Leg 201, a method was developed to semiquantify the pore water color. To provide a comparison to the Peru margin sites, a selected set of samples from Site 1244 was analyzed for yellowness relative to the JWBL standard used during Leg 201 (D'Hondt, Jørgensen, Miller, et al., 2003). The procedure involves a one to three dilution of pore water samples with deionized water and measurement of absorbance at 325 nm using the Milton Ray Spectronic 301 spectrophotometer.
Dissolved organic carbon (OC) analyses were conducted in a Shimadzu TOC-5000A total carbon analyzer. Approximately 1-5 mL of IW, depending on sample volume available, was placed in TOC vials, which had been prewashed with 10% HCL and combusted at 450°C for 4 hr. Samples were acidified to a pH of ~2 with 2-N HCL and placed in the ASI-5000A autosampler that was set up to purge the sample with purified air for 3 min prior to analysis. Triplicate injections of 25 µL were introduced to the combustion tube and heated to 680°C in the presence of a catalyst. The resulting CO2 was detected in a nondispersive IR gas analyzer (NDIR). Detector output was calibrated against potassium phthalate standard solutions covering a range of 0-49 ppm carbon.
Sodium and potassium were measured using a Dionex DX-120 ion chromatograph; whereas magnesium and calcium were measured by ICP-AES using a Jobin Yvon JY2000 spectrometer with yttrium as an internal standard (Mix, Tiedemann, Blum, et al., 2003). For all major cations, we used dilutions of IAPSO standard seawater as calibration standards. ICP-AES techniques were also used for the minor elements Mn2+, Fe2+, B, Sr2+, Ba2+, and Li+ (modified from Murray et al., 2000) by preparing calibration standards in an acidified (2.5% HNO3 by volume) sodium chloride matrix (35 g NaCl per liter). In addition, a 2.5% HNO3 matrix solution with yttrium at 10 ppm (1:10) served as an internal standard to dilute standards and acidified IW samples (Mix, Tiedemann, Blum, et al., 2003).
The reproducibility of techniques, expressed as 1-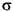 relative standard deviations of means, was evaluated by replicate analyses of calibration standards, of IAPSO, and/or of samples both within a given analytical run and in different analytical runs (Table T2). Chemical data for IW are reported in molar concentration units.
relative standard deviations of means, was evaluated by replicate analyses of calibration standards, of IAPSO, and/or of samples both within a given analytical run and in different analytical runs (Table T2). Chemical data for IW are reported in molar concentration units.
![]()