METHODS
We sampled 5-cm long quarter-round sections from eight stratigraphic horizons from Sections 208-1263A-33H-2, 33H-CC, and 34X-1 spanning the Paleocene/Eocene (P/E) boundary. The samples were freeze-dried at Kanazawa University (Japan). For samples below Section 208-1263A-34X-1, we freeze-dried three sediments after squeezing pore water on board the ship. The samples were scraped to remove surface contaminants. All 11 samples were ground and extracted with dichloromethane in soxhlet extractors for 48 hr. Each extract was dried under N2 flow, dissolved in 100 mL of hexane, and separated into lipid classes (saturated and unsaturated hydrocarbons, aromatic hydrocarbons, ketones, and polar compounds) by silica gel (deactivated with 5% H2O w/w) column chromatography.
We analyzed the saturated hydrocarbons using a Hewlett Packard 6890 gas chromatograph (GC) equipped with an on-column injector and flame ionization detector. The first fraction of samples from Core 208-1263A-34X contained unresolved complex mixtures (UCMs) over the region of n-alkanes C25–C33 on gas chromatograms. The urea adduction technique was employed to separate n-alkanes from the UCMs. Samples showing no obstructing UCMs for GC isotope ratio mass spectrometry (IRMS) analysis (Fig. F1A, F1B) were not treated. The urea adduction appeared to have no effect on the carbon isotope values of individual n-alkanes (Ellis and Fincannon, 1998).
We applied a saturated solution of urea in methanol to the first hydrocarbon fractions dissolved in the hexane:acetone (2:1) mixture. The mixture with white urea precipitate was shaken, settled 1 hr, and centrifuged. We separated the urea precipitate by pipetting the solvent and rinsing the urea three times with clean solvent to completely remove nonadducted materials. We dried the urea crystals under N2 flow and released the adducted compounds by addition of H2O to the urea crystals. After dissolution of urea in H2O, we added hexane to recover adducted compounds. The solvent including nonadducted compounds was dried under N2, and H2O was added to dissolve residual urea. Nonadducted compounds were recovered with hexane, and the urea adduction was repeated on the nonadducted compounds to achieve better recovery of n-alkanes. See the
"Appendix" for more information about the urea adduction technique.
We determined the carbon isotope values of n-alkanes C29 and C31 using a GC-IRMS system at Biogeochemical Laboratories, Indiana University. The GC-IRMS system employed a Hewlett Packard 5890 gas chromatograph equipped with a cool on-column injector and interfaced with a Finnigan MAT 252 mass spectrometer by combustion furnace (850°C). Samples were separated on a CP-SIL 8 CB low-breed/MS fused silica capillary column (50 m x 0.32 mm inner diameter with 0.4 mm film thickness). We used helium as carrier gas and programmed the GC oven to heat from 50° to 320°C at 4°C/min, followed by an isothermal hold at 320°C for 25 min. The carbon isotope values for n-alkanes C29, C31, and C33 were evaluated by a series of coinjected, deuterated n-alkanes of known carbon-isotopic composition (C20D42, C24D50, and C36D74). All carbon isotope ratios (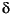 13C) are expressed as permil (
13C) are expressed as permil (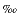 ) relative to the Vienna Peedee belemnite (VPDB) standard. Reported isotope ratios represent averaged values of duplicate analyses. Instrumental standard deviation for the analysis (1
) relative to the Vienna Peedee belemnite (VPDB) standard. Reported isotope ratios represent averaged values of duplicate analyses. Instrumental standard deviation for the analysis (1 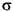 ) is <0.3 based on repeated analysis of isotopically known n-alkane laboratory standards (C29, C31, and C33).
) is <0.3 based on repeated analysis of isotopically known n-alkane laboratory standards (C29, C31, and C33).
