DISCUSSION
Precipitation
of Carbonates
Mixing of sulfate- and
methane-bearing fluids is occurring at Site 996 (Paull, Matsumoto, Wallace, et
al., 1996) and other vent locations (Suess and Whiticar, 1989; Borowski et al.,
1996; Wallmann et al., 1997). Sulfate gradients with sulfate concentrations
approaching 0 mM near the seafloor (0.10-6.95 mbsf; Paull, Matsumoto, Wallace,
et al., 1996) suggest that sulfate depletion is driven by methane flux from
below (Borowski et al., 1996) and that anaerobic methane oxidation is the
principal sulfate-consuming process at Site 996. The upward methane flux, which
is required to sustain these reactions above the Blake Ridge Diapir, is most
likely occurring along fluid conduits that were observed in seismic reflection
data and in the form of vertical, gas hydrate-cemented veins in the sedimentary
section.
The authigenic carbonate
cements at Site 996 are primarily composed of aragonite (Table
1; Fig. 5), which is
consistent with authigenic aragonite formation at other vent sites (e.g.,
Hovland et al., 1987; Matsumoto, 1990; Jørgensen, 1992; Savard et al., 1996).
Temperature, the degree of pore-fluid supersaturation, and the presence of oxic
or anoxic conditions seem to be the major controlling factors for the
precipitation of different carbonate minerals (Burton and Walter, 1987; Hovland
et al., 1987; Burton, 1993). The relative importance of those mechanisms,
however, is still poorly understood. Elevated temperatures generally seem to
favor aragonite precipitation, whereas changes in saturation state seem to have
little effect. There have been no definitive means (petrographic, isotopic, or
geochemical) for distinguishing between carbonate cements formed under oxic or
anoxic conditions. Although aragonite formation has generally been associated
with oxic environments at or near the seafloor (Longman, 1980; Hovland et al.,
1987, Matsumoto, 1990), the co-occurrence of aragonite and authigenic pyrite in
almost all Site 996 samples (Fig. 3F)
is difficult to reconcile with oxic conditions, because pyrite can only form in
anoxic, sulfate-reducing environments. Thus, the aragonite cements at Site 996
must have formed very near the sediment/water interface in an oxygen-depleted
environment where the generation of HCO3- by anaerobic
methane oxidation resulted in oversaturation with respect to aragonite.
The darker rims on many of
the intraclasts may be dissolution rinds that contain less CaCO3. The
interaction of sulfide-rich pore fluids with oxygenated seawater (HS-
+ 2O2 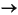 SO4-2
+ H+) produces locally acidic water (Paull and Neumann, 1987),
generating dissolution in carbonates at the sediment/water interface. The rough
and partially dissolved surfaces of aragonitic bivalve shells also indicate
exposure (either syn- or postdepositionally) to a locally corrosive environment
at the seafloor (Fig. 4C). The
complex juxtaposition of dissolution surfaces with cemented sediment suggests
that the position of the oxic-anoxic interface may fluctuate over time, perhaps
in response to variations in the flux of methane from below.
SO4-2
+ H+) produces locally acidic water (Paull and Neumann, 1987),
generating dissolution in carbonates at the sediment/water interface. The rough
and partially dissolved surfaces of aragonitic bivalve shells also indicate
exposure (either syn- or postdepositionally) to a locally corrosive environment
at the seafloor (Fig. 4C). The
complex juxtaposition of dissolution surfaces with cemented sediment suggests
that the position of the oxic-anoxic interface may fluctuate over time, perhaps
in response to variations in the flux of methane from below.
Controls on
the Geochemical and Isotopic Composition of the Authigenic Carbonates
The depletion in 13C
of the authigenic carbonates at Site 996 (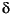 13C
as low as -48
13C
as low as -48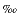 ) indicates that CO2
derived from methane oxidation was the primary carbon source for these
carbonates. Based on methane 13C
values of -62 to -72
at Site 996 (Paull et al., Chap.
7, this volume), we estimate that up to 75% of the carbon incorporated
into authigenic carbonates at this site was derived from biogenic methane.
) indicates that CO2
derived from methane oxidation was the primary carbon source for these
carbonates. Based on methane 13C
values of -62 to -72
at Site 996 (Paull et al., Chap.
7, this volume), we estimate that up to 75% of the carbon incorporated
into authigenic carbonates at this site was derived from biogenic methane.
Measurements of the oxygen
isotopic composition of pore water of near surface sediments above the Blake
Ridge Diapir revealed 18O
values of 0.1 to 0.9
with a mean of 0.3 (Borowski et al., 1997).
Using the 18Oaragonite/T
relationship established by Hudson and Anderson (1989), the 18O
values of the aragonite cements indicate a precipitation temperature of about
4ºC. This is in good agreement with the observed bottom-water temperature of
3.5ºC at Site 996 (Paull, Matsumoto, Wallace, et al., 1996). It is therefore
reasonable to infer that these cements precipitated in, or near, isotopic
equilibrium with the regional bottom water.
Carbon
Isotopic Composition of Pore Fluids and Authigenic Carbonates
The carbon isotopic
composition of 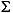 CO2 in
near-surface sediments at Site 996 varies between -28
and -45 (Paull et al., Chap.
7, this volume). Such negative 13C
values are typical for concurrent microbial-mediated oxidation of sedimentary
organic matter and methane originating from depth (Suess and Whiticar, 1989). As
indicated by 13C
values of -62 to -72,
methane that is consumed at Site 996 is predominately biogenic in origin (Paull
et al., Chap. 7,
this volume). As authigenic carbonates derive their carbon from the pore-water CO2
pool (Suess and Whiticar, 1989), the similarity between the 13C
values of the carbonates (-30.5 to -48.4)
throughout the sedimentary section and pore-water CO2
(-28 to -45)
at shallow depth indicates that the carbonates have formed from this shallow CO2
pool. Furthermore, carbon isotope values of CO2
at Site 996 are most negative near the sediment/water interface and become more
positive with depth (Fig. 8).
The 13C
values of recovered authigenic carbonates, however, do not show such a trend (Fig.
9). This dissimilarity again suggests that carbonate nodules recovered
from greater depth probably formed near the seafloor, from a 13C-depleted
carbon pool similar to the one that currently exists.
CO2 in
near-surface sediments at Site 996 varies between -28
and -45 (Paull et al., Chap.
7, this volume). Such negative 13C
values are typical for concurrent microbial-mediated oxidation of sedimentary
organic matter and methane originating from depth (Suess and Whiticar, 1989). As
indicated by 13C
values of -62 to -72,
methane that is consumed at Site 996 is predominately biogenic in origin (Paull
et al., Chap. 7,
this volume). As authigenic carbonates derive their carbon from the pore-water CO2
pool (Suess and Whiticar, 1989), the similarity between the 13C
values of the carbonates (-30.5 to -48.4)
throughout the sedimentary section and pore-water CO2
(-28 to -45)
at shallow depth indicates that the carbonates have formed from this shallow CO2
pool. Furthermore, carbon isotope values of CO2
at Site 996 are most negative near the sediment/water interface and become more
positive with depth (Fig. 8).
The 13C
values of recovered authigenic carbonates, however, do not show such a trend (Fig.
9). This dissimilarity again suggests that carbonate nodules recovered
from greater depth probably formed near the seafloor, from a 13C-depleted
carbon pool similar to the one that currently exists.
There are also indications
that the carbon isotopic composition of the CO2
pool at any one spot changes over time. An isotopic profile of 13C
values across an aragonite-cemented vein within one of the carbonate nodules (Fig.
10) shows a symmetric trend towards more 13C-depleted carbon
isotope values from the rim to the center of the vein. This trend can be
explained as the result of a shift in the methane pool from less 13C-depleted
to strongly 13C-depleted methane, possibly in response to changes in
flow rate (Suess and Whiticar, 1989) or recycling of light carbon in the shallow
methanogenic zone (Borowski et al., 1997). Alternatively, this may reflect local
changes in the relative importance of the different carbon sources to the pore
fluids.
Oxygen
Isotopic Variations
As a first approximation, 18O
values of the carbonate nodules indicate that they precipitated in or near
isotopic equilibrium with the regional bottom water. Closer inspection, however,
shows that the oxygen isotopic composition of carbonates recovered from shallow
depth (0-3 mbsf) differs from those recovered from deeper in the sediment (Fig.
6, Fig. 9). This difference in
the oxygen isotopic composition is roughly 1 (Fig.
6). Although currently highly speculative, this difference in 18O
might be related to changes in bottom-water conditions, corresponding to glacial
and interglacial time periods. However, the relatively poor depth and age
control on the carbonate precipitates does not allow a detailed comparison to
established Pleistocene 18O
records.
Strontium
Isotope Ratios
Secular variations in the
strontium isotopic composition of seawater are well known (e.g., Elderfield,
1986; Farrell et al., 1995). The strontium isotopic composition of carbonate
minerals will reflect the strontium isotopic composition of the water in which
they formed (Hess et al., 1986). Thus, strontium isotopes can be used to
constrain the source of fluids for authigenic carbonate precipitation in marine
sediments (Sample and Reid, 1998). To evaluate the source of fluids at this
site, we compared the 87Sr/86Sr values of authigenic
carbonates to the 87Sr/86Sr values of pore fluids in
nearby sedimentary sections not influenced by venting fluids (Sites 994, 995,
and 997).
Because all of the samples
that were recovered from Site 996 are Pleistocene in age (Paull, Matsumoto,
Wallace, et al., 1996), only a nominal variation in the Sr isotopic composition
of the authigenic carbonates is to be expected if the carbonates have formed at
or near the seafloor in contact with Pleistocene bottom waters. If, however,
there has been addition of strontium from depth, one would expect a different Sr
isotopic composition because pore waters from greater depth are known to vary in
this area. At Sites 994, 995, and 997 on the Blake Ridge, the Sr isotopic
composition at and below the base of gas hydrate stability ranges from 0.709006
to 0.709043 (Fig. 7). If the
source of the Sr captured in the authigenic carbonates was being carried in
fluids moving upward along a fault above the Blake Ridge Diapir, the 87Sr/86Sr
values should reflect this deeper pool. At Site 996, however, pore-water Sr
values are clearly indistinguishable from measurements of modern (0.709175) and
contemporaneous seawater (Farrell et al., 1995). A comparison with Farrell's
seawater curve (Fig. 7) also
shows that the 87Sr/86Sr values of carbonate precipitates
recovered throughout the sedimentary section are approximately consistent with
the expected 87Sr/86Sr composition based on the age of
their host sediment. Thus, our data do not indicate an addition of Sr from
deep-seated fluids at Site 996.
The Dolomite
Nodule
A single dolomite nodule,
with a fabric similar to our "type 3" authigenic carbonate (Fig.
3E), was recovered from 51.6 mbsf in Hole 996E. Dolomite (composed of 45
mol% Mg) occurs as microcrystalline cement. The isotopic characteristics of this
nodule suggest an origin distinct from the rest of the authigenic carbonates in
this section. The 13C
values of the dolomite nodule (-13.1 to -19.2)
are significantly more enriched in 13C than those of authigenic
aragonite recovered elsewhere in the sedimentary section. Furthermore, the 18O
values (4.8-5.4)
indicate a temperature of formation significantly higher (18º-24ºC, using the 18O/temperature
relation of Northrop and Clayton [1966]), than those of the other authigenic
carbonates at this site. Although we could assume that the dolomite nodule
formed from 18O-depleted water (estimated to be -4.0
to -4.5 standard mean ocean water [SMOW],
based on Northrop and Clayton, [1966]), oxygen isotope measurements of pore
water at Site 996 (Egeberg, Chap.
22, this volume) do not indicate such light 18O
values.
Although authigenic
dolomites have been described from various vent locations (e.g., Matsumoto,
1990; Jørgensen, 1992; Kopf et al., 1995), we do not know at this point if the
dolomite nodule at Site 996 is directly related to the venting process. The
isotopic similarity of the nodule to dolomite found elsewhere on the Blake Ridge
at Sites 994, 995, and 997 (Rodriguez et al., Chap.
30, this volume), and the fact that it is the only sample of its kind at
Site 996, suggests an origin independent of fluid venting.
CaCO3
Precipitation at Site 996
Our data indicate that
precipitation of authigenic carbonates at this site took place at (or near) the
seafloor and that continued carbonate formation with increasing sediment depth
has not occurred. This inference is supported by the observation that
precipitates do not show any significant petrographic or mineralogical changes
with depth and that aragonite is the primary authigenic carbonate at Site 996.
In addition, both microscopy and electron microprobe analysis do not provide
evidence for multistage cementation or changes in the mineral chemistry with
depth (mineralogical changes would be expected if nodules grew, or continued to
grow, at several depth intervals). Furthermore, 13C
values of the authigenic carbonates (-30.5 to
-48.4) are similar to the carbon isotopic
composition of pore water CO2 at
shallow depth (-28 to -45).
At greater depths, 13C
values of CO2 become
progressively 13C enriched (+10.5
at 43 mbsf), demonstrating that the authigenic carbonates could not have formed
in equilibrium with bicarbonate at ambient depth. If these observations can be
extrapolated to other vent sites, precipitation of authigenic carbonate at vent
sites takes place only at or near the sediment/water interface, and the fluid
conduit itself will not be distinctly marked. This means that only the seafloor
expression of vent sites will be preserved (as carbonate crusts) and that fossil
fluid conduits will be difficult to find in the sedimentary record.
The occurrence of
authigenic carbonates at several depth horizons, therefore, can be used to
estimate the duration, and lateral shifting, of fluid venting above the Blake
Ridge Diapir. Given that the deepest aragonite nodule was recovered from about
30 mbsf, and assuming a constant sedimentation rate of ~48 m/m.y. (Paull,
Matsumoto, Wallace, et al., 1996), we can infer that seepage has been going on
in this area for at least 600,000 yr. Variations in the oxygen isotopic
composition (which might be related to glacial and interglacial periods) support
this inference.
