RESULTS AND DISCUSSION
ODP labels, depths, ages, and the bulk geochemical properties with their corresponding mass accumulation rates for each sample are given in Table T1.
The hydrocarbons eluted in Fraction 1 and their distributions are illustrated in the gas chromatograms in Figure F1. Identities and quantities of the compounds corresponding to the numbered peaks are listed in Table T2. The dominant series of compounds in both samples are n-alkanes with a bimodal distribution (Fig. F2A) biased toward higher chain lengths (n-C25 to n-C35). Even-number homologues of the longer chain n-alkanes are conspicuously absent in any detectable amount. Such an odd-over-even predominance is indicative of a terrestrial higher plant origin (Eglinton et al., 1962). The source of this terrigenous signal is likely to be continental Africa through eolian transport owing to the trajectory of the dominant along-shore trade winds and the absence of any major fluvial input directly affecting Site 1084 (Gagosian et al., 1981, 1987; Simoneit et al., 1977). The higher abundances of n-alkanes in the near-surface sample relative to the mid-Pliocene sample could be a result of increased trade wind transport of eolian material. However, such an interpretation is complicated by the aridification and expansion of the Namib Desert associated with the intensification of upwelling leading to a decline in the abundance of terrestrial higher plants (Dowsett and Willard, 1996). The contribution of n-alkanes to the total organic matter (as mass per milligram of Corg) (Fig. F2B) is similar for each sample, indicating that the input of terrigenous organic carbon to the site may not have increased coincident with the increase in trade wind strength.
Pristane (Pr) and phytane (Ph) are present in both samples, with a Pr:Ph ratio of <1 (0.95 mbsf = 0.77 and 579.92 mbsf = 0.54). Studies have suggested that values less than unity may indicate anoxic phytol diagenesis (Didyk et al., 1978). However, various factors restrict this interpretation (ten Haven et al., 1987), and the ratio may have been influenced by direct bacterial inputs (Han and Calvin, 1969; Risatti et al., 1984). A C25 isoprenoid (2,6,10,15,19-pentamethyleicosane) is also present in both samples. This compound is a biomarker for methanogenic bacteria (Brassell et al., 1981) and was identified in sediments underlying the upwelling system off northwest Africa (ten Haven et al., 1989). The series of 3-methyl and 5-methyl branched alkanes found in the surface sample may also be indicative of an archaebacterial input (Brassell et al., 1981; Kenig et al., 1995; Shiea et al., 1990). An unsaturated compound in this series and monounsaturated isoprenoids (pristene and phytene isomers) in the deeper sample suggests that the depositional environment may have been less reducing than for the surface sample. Moreover, the presence of these compounds in the deep sample suggests that thermal maturation has not progressed significantly at depth in Hole 1084A (Van Graas et al., 1981). No compounds from the series of highly branched isoprenoid (HBI) alkanes or alkenes were identified in either sample. It is possible that the HBI compounds are present in low concentrations and/or coelute with other compounds. These compounds have been identified in several marine diatom species (Belt et al., 1996; Johns et al., 1999; Sinninghe Damsté et al., 1999; Volkman et al., 1994; Wraige et al., 1997) and in a variety of marine sediments (see Rowland and Robson, 1990, for a review). Their absence in Hole 1084A sediments is conspicuous owing to micropaleontological evidence for large inputs of diatoms (Lange et al., 1999). Sulfur incorporation has been shown to be a rapid mode of diagenetic removal of the HBI alkenes (Kohnen et al., 1990; Sinninghe Damsté et al., 1989a), but their solvent extractable diagenetic products appear to be absent from both samples (other organic sulfur compounds are discussed below). The absence of HBI compounds was also noted in the diatomaceous sediments underlying the coastal upwelling regime of the Peru margin (ten Haven et al., 1990) and the central Arabian Sea (Prahl et al., 2000). This latter study showed that significant quantities of HBIs were exported from the water column, but they were not detected in the underlying surface sediments. However, Schouten et al. (2000) have reported a series of HBI polyenes in surface samples from the Arabian Sea although absolute quantities were not given.
The relative abundance of steroidal hydrocarbons to n-alkanes increases in the deeper sample, indicating a depth/time-dependent transformation from their sterol and/or steroidal ketone precursors (Mackenzie et al. 1982; Peakman and Maxwell, 1988; Brassell et al., 1984). Limited double-bond isomerization to ster-4-enes and ster-5-enes suggests that the sediments have undergone diagenetic transformation. The absence of fully saturated steranes indicates that the sediments remain thermally immature at depth (Gagosian and Farrington, 1978; ten Haven et al., 1989). Unsaturated and 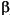 isomers of the bacterially derived hopanoidal hydrocarbons (Ourisson et al., 1984) are present in both samples as well as the diagenetically rearranged
isomers of the bacterially derived hopanoidal hydrocarbons (Ourisson et al., 1984) are present in both samples as well as the diagenetically rearranged 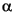 / isomers. The distribution of these hydrocarbons per gram of dry sediment and per gram of Corg does not indicate any clear trend toward increasing thermal maturity with depth (Ensminger et al., 1977; Brassell and Eglinton, 1983; Farrimond et al., 1986a). The distribution of fernene isomers (also of bacterial origin) between the surface and deep samples is similar to that observed in the upwelling sediments off northwest Africa (ten Haven et al., 1989). The distribution of fernenes appears to follow progressive isomerization from the
/ isomers. The distribution of these hydrocarbons per gram of dry sediment and per gram of Corg does not indicate any clear trend toward increasing thermal maturity with depth (Ensminger et al., 1977; Brassell and Eglinton, 1983; Farrimond et al., 1986a). The distribution of fernene isomers (also of bacterial origin) between the surface and deep samples is similar to that observed in the upwelling sediments off northwest Africa (ten Haven et al., 1989). The distribution of fernenes appears to follow progressive isomerization from the 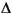 7 to the thermodynamically more stable 8 and 9(11) isomers with depth.
7 to the thermodynamically more stable 8 and 9(11) isomers with depth.
Two C20 isoprenoid thiophenes were identified in Fraction 1 of both samples (Table T2). These compounds have been found in many marine sediments, including previous studies form the Walvis Ridge DSDP studies (Brassell et al., 1986b; ten Haven et al., 1992). The thiophenes are formed during the early stages of diagenesis in the water-column and surface sediments (Brassell et al., 1986b; Kohnen et al., 1991a). Their biological precursors are likely to be phytenyl moieties that react with reduced inorganic sulfur species (H2S and hydrogen polysulfides) to form the sulfur incorporated thiophenes (Brassell et al., 1986a; Sinninghe Damsté et al., 1989b). The greater abundance of the thiophenes in the deeper sample suggests a more anoxic/reducing environment during deposition. This interpretation contradicts the information on depositional setting given by the presence of the unsaturated hydrocarbons in the deep sample. However, interpretation of the distribution of the thiophenes in the solvent extractable fraction is complicated by the potentially more significant form of sulfurization via sulfur bridges into the bound macromolecular fraction (Kohnen et al. 1991a, 1991b, 1991c).
Wax Esters and Sterol Ethers/Esters
Fraction 2 was dominated by high molecular-weight compounds that were tentatively identified from their relative GC retention times and mass spectra as wax esters and compounds containing sterol moieties probably as alkyl ethers and/or esters (Fig. F3). The exact structures of the sterol compounds were difficult to identify owing to the absence of a significant molecular ion in their electron impact mass spectra. Such compounds have been identified in sediments deposited under the upwelling cells of Walvis Bay (Boon and de Leeuw, 1979), the Peru margin (ten Haven et al., 1990), and the Arabian Sea (Schouten et al., 2000). The source of these wax and sterol ethers/esters has been assigned to zooplankton grazing on algal sterols (Wakeham, 1982). However, the noticeable occurrence of these compounds in sediments underlying waters with high abundances of diatoms and the disparity between the sterol moiety of the ethers compared to the free sterols studied by Schouten et al. (2000) has led these authors to suggest that diatoms may be the direct source of sterol ethers. Further analyses using chemical ionization mass spectrometry (Lusby et al., 1984) and saponification are required for full structural characterization. The highest abundances of the ethers/esters are in the surface sample, including the long-chain (C37 and C38) alkyl alkenoates (see "Long-Chain Alkenones and
Alkenoates" below). The presence of a series of unknown compounds eluting prior to the wax/sterol esters in Fraction 2 was noted. These compounds are dominant in the deep sample. Their mass spectra contain a large base peak at m/z 231, with no apparent molecular ion or any other characteristic fragment ions.
Steroidal/Hopanoidal Ketones
Uncharacterized ketone-substituted steroid and hopanoid compounds eluted in Fraction 3 (Fig. F4). Steroid ketones have previously been identified in the sediments from the BCS (Gagosian and Smith, 1979), where their origin was suggested to be a diagenetic intermediate in the microbiologically/chemically mediated degradation of stenols to sterenes. However, a direct input from dinoflagellates is also possible (Harvey et al., 1988).
Long-Chain Alkenones and Alkenoates
The long-chain alkenones are ubiquitous in marine sediments (Brassell, 1993) and have been found to be among the largest components of the extractable lipids in upwelling sediments (ten Haven et al., 1989, 1990; Hinrichs et al., 1999; Farrimond et al., 1990). These compounds are the most abundant lipids in Fraction 3 (Fig. F4) and have concentrations and accumulation rates considerably higher than any other compound in any of the fractions (Table T3). The C37-C39 alkenones and alkenoates are biomarkers for the haptophyte algae (Volkman et al., 1980; Marlowe et al., 1984; Conte et al., 1994), notably the ubiquitous coccolithophorids Emiliania huxleyi and Gephyrocapsa oceanica. In this respect, it would appear that haptophyte algae have been dominant members of the plankton at Site 1084 throughout the past 4.5 m.y. (Marlow et al., 2000). The highest accumulation rate is found in the surface sample (1.8 mg/cm2/k.y. compared to 0.6 mg/cm2/k.y. in the deep sample). The concentration of alkenones relative to Corg becomes enriched in the deep sample (1.44 mg/g Corg compared to 0.97 mg/g Corg in the surface sample) as a likely result of the preferential preservation of the alkenones relative to other more labile organic compounds. The observation that alkenone abundances are significant in a variety of marine sediments predating the first occurrence of E. huxleyi at 268 ka (Thierstein et al., 1977) suggests that alkenones have been biosynthesized by phylogenetic ancestors throughout the late Neogene and possibly earlier (Emeis et al., 1995; Dzvonik, 1996; Farrimond et al., 1986b; Herbert and Schuffert, 1998; Marlowe et al., 1990; Müller et al., 1997; Rullkötter et al., 1998). The C40 di-unsaturated alkenone was tentatively identified in the deep sample, as the molecular ion was particularly weak. Full characterization will be possible with chemical (NH3) ionization mass spectrometry (Rosell-Melé et al., 1995a). This compound has previously been reported in Cretaceous black shales (Farrimond et al., 1986b). The di- and tri-unsaturated C37 alkenones (37:2 Me and 37:3 Me) are commonly used for reconstructing paleo-sea surface temperatures (SST) owing to the observation that the ratio of 37:2 Me/(37:2 Me + 37:3 Me) = UK37´ has a linear relationship with growth temperature and can be accurately calibrated to the SST through core-top studies and cultures (Brassell et al., 1986a; Müller et al., 1998; Prahl and Wakeham, 1987; Prahl et al., 1988; Rosell-Melé et al., 1995b). The UK37´ and SST estimates for the two samples are given in Table T3. The UK37´ value for the surface sample corresponds to the cooling interval following the thermal optimum of marine oxygen isotope Stage 5 (Marlow et al., 2000). Interpretation of a complete time series of UK37´-derived SST for the 4.5-m.y. period between the surface and deep sample reported here suggests that the elevated SST for the deep sample probably reflects a combination of increased global temperatures and less intense upwelling relative to the late Pleistocene (Marlow et al., 2000).
Long-Chain Alkyl-1,n-diols and Alkyl-1-ol-n-ones
Mid-chain (1,15) C30-C32 diols and keto-ols were first reported in Black Sea sediments, and their source was postulated to be of cyanobacterial origin (de Leeuw et al., 1981). A series of alkyl diols were subsequently identified in marine and freshwater eustigmatophyte algae (Volkman et al., 1992; Volkman et al., 1999). However, compositional differences between the positional isomers found in cultures and sediments suggest that eustigmatophyte algae are not the major source of these compounds and the source organisms are still as yet unknown (Versteegh et al., 1997). Contrary to other published results for upwelling sediments (ten Haven et al., 1992; McCaffrey et al., 1991), including BCS sediments (Hinrichs et al., 1999), alkyl diols were not readily identifiable in either of the samples. However, a series of alkyl keto-ols were easily identifiable in large quantities from their mass spectra, with large fragment ions at m/z = 130, 143, and M+-15. The surface sample contains a significantly larger quantity of keto-ols, a wider diversity of chain lengths, and a monounsaturated compound. The distribution of the 1,15-C30 keto-ol and 1,15-C32 keto-ol in the surface sample may be related to the surface water conditions at the time of production (Versteegh et al., 2000). The normalized ratio of the two saturated ket-ols defined by Versteegh et al. (1997) as [100 × (1,15-C30 keto-ol)]/[([1,15-C30 keto-ol) + (1,15-C32 keto-ol)] is ~72%. This ratio is similar to that found by Versteegh et al. (2000) in sediments from the Southern Angola Basin (11°35´S, 11°41´E) corresponding to the same time interval and these authors speculate a relationship to surface water salinity. Owing to the possible compound-environment relationship for the keto-ols, it is difficult to determine whether the different distributions and absolute quantities between the surface and deep samples are caused by diagenetic alteration or changes in the surface water conditions.
Straight-chain alcohols and acids eluted in Fraction 4 (Fig. F5). Three even-chain-length alcohols (C18, C22, and C26) were present in the surface sample, but only the C18 homologue was found in the deep sample. An even-over-odd predominance was also reported in Peru margin upwelling sediments (Farrimond et al., 1990), implying an input derived from higher plants (Kolattukudy, 1976; Gagosian et al., 1981, 1987) most probably through eolian transport.
Fatty acids formed a significant fraction of the polar compounds (Table T4), with the surface sample having the largest overall abundance. An absolute quantitative interpretation of the fatty acid distribution is limited by the absence of an acidification step during the preparative procedures. The distribution of fatty acids in both samples is bimodal with maxima at C16 and C26 (Fig. F6). However, the dominant maxima are different for each sample, with the surface sample biased toward longer chain lengths and the deep sample biased toward shorter chain lengths with a uniformly low abundance of long-chain fatty acids. A C18:1 mono-unsaturated acid was identified in both samples. The biological source of fatty acids is varied (Volkman et al., 1998; Gagosian et al., 1981, 1987). Fatty acids at Site 1084 are most likely derived from a mixture of autochthonous inputs from microalgae and bacteria and to a lesser extent from eolian transport. Similar distributions have been described for other late Quaternary sediments from the BCS (Hinrichs et al., 1999).
Sterols (C27-C30) eluted in Fraction 4 (Fig. F5) and were identified from their relative retention times and comparison with published mass spectra (Brassell, 1980; McEvoy, 1983). All sterols are significantly more abundant per gram Corg in the surface sample relative to the deep sample (Table T4) as a possible result of the progression of diagenetic transformation in the deep sample and from increasing productivity following the intensification of upwelling since the mid-Pliocene. The interpretation of sterols as source indicators for surface BCS sediments is complicated by strong diagenetic degradation during transport through the oxygenated water column (Hinrichs et al., 1999).
The 4-desmethyl C27 sterols cholest-5-en-3-ol and 5-cholestan-3-ol are present in both samples, with the former being the most abundant single sterol in the deep sample. The stanol can have both a biological (e.g., dinoflagellates) and diagenetic (from the unsaturated sterol) origin (Robinson et al., 1984; Mackenzie et al., 1982). The C28 sterols 24-methylcholest-5,22-dien-3-ol and 24-methy-5-l-cholest-22-dien-3-ol are present in both samples. The unsaturated compound is found in diatoms, haptophytes, and cryptophytes (Goad et al., 1983; Volkman, 1986; Volkman et al., 1998). The C29 sterols are difficult to interpret as source markers owing to their presence in both marine algae and higher plants (Volkman et al., 1998), although a marine origin is most likely in this case (e.g., diatoms and raphidophytes). The C29 sterols are the most abundant sterols in the surface sample but are depleted in the deep sample. A similar observation in BCS sediments was interpreted as diagenetic loss and/or reduced biological supply (Hinrichs et al., 1999). C30 sterols were present in both samples, including the 4-methylsterols 4,23,24-trimethyl-5-cholest-22-en-3-ol (dinosterol) and 4,23,24-trimethyl-5-cholest-8(14)-en-3-ol, which have been used as biomarkers for dinoflagellates (Boon et al., 1979; Robinson et al., 1984).
