MATERIAL AND METHODS
During Ocean Drilling Program (ODP) Leg 185 at Site 1149 (Holes 1149A and 1149B), we recovered 180 m of sediments (lithologic Unit I and Subunits IIA and IIB) from the northwest Pacific, at 31°20.519´N, 143°21.078´E (Hole 1149A; 5818 m water depth) and at 31°20.532´N, 143°21.060´E (Hole 1149B; 5818 m water depth) (Fig. F1). Unit I (0-118.2 meters below seafloor [mbsf]) is composed of argillaceous hemipelagic sediments (late Miocene-late Pliocene in age), including many volcanic glass fragments, feldspar grains, and siliceous biogenic tests. Subunit IIA (118.2-149.5 mbsf) is characterized by brown pelagic clay, including small amounts of volcanic glass fragments and siliceous biogenic tests. Subunit IIB (149.5-180 mbsf) is composed of dominantly brown pelagic clay. The lowermost part of Subunit IIB includes zeolitic (clinoptilolite) pelagic clay below 175 mbsf. The age of Unit II is unknown because of the lack of index fossils, but comparable lithology at Deep Sea Drilling Project (DSDP) Leg 20, Hole 196 (30°1.162´N, 148°5.748´E; 6194 m water depth), ~500 km east of Site 1149, is considered to be Late Cretaceous in age (Shipboard Scientific Party, 2000). Below Subunit IIB, Cretaceous chert and marlstone continue down to ~400 mbsf, above basaltic rocks.
Tube samples (each ~3 cm diameter, 5 cm in length), two samples per each core, were packed on board with certain orientation in cylindric plastic cases to keep them wet until they were divided into SEM and AMS samples on shore.
Since the grain density, porosity, and void ratio of Subunit IIB from Hole 1149A were not measured during Leg 185, we measured grain density (g/cm3) (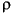 g) of four samples from Subunit IIB (Samples 185-1149A-18H-2, 65-67 cm; 18H-4, 75-77 cm; 19X-1, 40-42 cm; and 20X-1, 88-90 cm) using a pycnometer. The porosity (
g) of four samples from Subunit IIB (Samples 185-1149A-18H-2, 65-67 cm; 18H-4, 75-77 cm; 19X-1, 40-42 cm; and 20X-1, 88-90 cm) using a pycnometer. The porosity (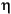 ) and void ratio (e) were calculated from the above grain density, the water content by dry weight (W), and the dry bulk density (g/cm3) (d) reported by Shipboard Scientific Party (2000) as follows:
) and void ratio (e) were calculated from the above grain density, the water content by dry weight (W), and the dry bulk density (g/cm3) (d) reported by Shipboard Scientific Party (2000) as follows:
Void ratio in Equation (1) (Lambe and Whiteman, 1969),
-
e = {[(1 + W)/d][gw]} - 1, (1)
and porosity,
-
= e/(1 + e),
(2)
where w = density of seawater (typical average value) = 1.024 g/cm3.
The AMS samples were obtained from the tube samples through the insertion of 7-cm3 plastic cubes in the right orientations. The remains of the samples were used for smear slide observations, grain-size analysis, and XRD analysis. AMS of the cube samples was measured using a AGICO KLY-3 magnetic susceptometer at a setting of 0.04 mT low magnetic field.
The AMS is geometrically represented by a magnetic susceptibility ellipsoid with three principal axes: the maximum (Kmax), intermediate (Kint), and minimum (Kmin) magnetic susceptibility. In general, the ellipsoid is controlled by arrangement of magnetic particles in the sediments (Tarling and Hrouda, 1993). The following parameters were used in this study to represent the shape of the magnetic susceptibility ellipsoid in Equations (3) (Jelinek, 1981), (4) (Stacey et al., 1960), and (5) (Basley and Buddington, 1960):
-
P´ = exp(SQR{2[(1 - m)2 + (2 - m)2 + (3 - m)2]}),
(3)
-
F = Kint/Kmin, and
(4)
-
L = Kmax/Kint,
(5)
where
-
1 = ln(Kmax);
-
2 = ln(Kint);
-
3 = ln(Kmin),
-
m = (1 + 2 + 3)/3;
-
P´ = corrected anisotropy degree;
-
F = foliation; and
-
L = lineation parameter.
The declinations of the Kmax, Kint, and Kmin of the AMS were corrected to magnetic north using the on board measured paleomagnetic data (Shipboard Scientific Party, 2000).
Because all the minerals in marine sediments contribute to the AMS in various degrees, it is important to define the kind of mineral most responsible for the measured magnetic fabric by the following two methods. Both vibrating sample magnetometer (VSM) measurements and SEM energy-dispersive spectrometer (EDS) analyses were used for this purpose. The VSM of Molspin Co. Ltd. measures susceptibility in high magnetic fields (Khf) of 500-1000 mT. The Khf is generally a result of magnetic minerals, mainly paramagnetic, not ferrimagnetic minerals (Housen and Sato, 1995; Housen, 1997). On the other hand, low field susceptibility (Klf) in the sediments measured at 0.04 mT by the KLY-3 can be subdivided into components that are contributed by both ferrimagnetic and paramagnetic minerals (Housen and Sato, 1995; Housen, 1997). The ratio of Khf/Klf is inversely proportional to the relative contribution extent of the ferrimagnetic minerals to the Klf (Housen and Sato, 1995; Housen, 1997).
Additionally, ferrimagnetic minerals were separated by a hand magnet from the sediment samples dispersed into distilled water in order to observe their individual shape. The collected ferrimagnetic minerals were attached to SEM stubs using carbon tape and then coated with carbon. Their chemical composition was analyzed using EDS.
The grain-size distribution was obtained using a CIRAS1064 laser diffraction grain-size analyzer. A wet sediment weight of ~0.1 g for each sample was dispersed into boiled water in a glass beaker and then left for 24 hr than was further dispersed by an ultrasonic vibrator for 30-60 s just before measurement.
The XRD analysis was conducted for samples finer than 5 µm diameter on a polished glass slide. Three slides were analyzed for each sample in this study; the first slide sample was untreated, the next one was boiled in a dilute HCl (6 N) solution for 1 hr, and the last one was treated with ethylene glycol. CuK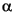 was used under 40-kV and 15-mA conditions by a RIGAKU RAC-A system.
was used under 40-kV and 15-mA conditions by a RIGAKU RAC-A system.
Using HCl treatment, the kaolinite (001) (7 Å) peak could be identified from the original doublet peaks of kaolinite (001) and chlorite (002) because chlorite in the samples was dissolved by this treatment. Through the ethylene glycol treatment, the smectite (001) (12 Å) peak was found shifted from the original doublet peaks of chlorite (001) and smectite (001) to 15 Å. From the relative extent of each peak area, the component ratio of clay minerals was calculated in each sample qualitatively (Oinuma, 1968; Aoki and Kohyama, 1998) as follows: smectite (15 Å) = 1.0, illite (10 Å) = 3.6, chlorite (7 Å) = 3.6, and kaolinite (7 Å) = 3.6.
Preparation of Samples
for SEM and Polarized Microscope
The SEM samples and thin section samples were prepared by the freeze-drying method (Takizawa et al., 1995) in order to avoid microfabric disturbance under the air-drying process that is due to the effect of surface tension of pore water. The sediment from Unit I that was subjected to the air-drying method suffered 25%-40% volume shrinkage, whereas that from Unit II suffered ~60%. As a result, the microfabrics in both units were completely distorted (Fig. F2).
The freeze-drying method was conducted as follows. First, the pore water in the sediment was replaced by ethanol and then by t-butyl alcohol for several months. Next, t-butyl alcohol in the pores was quickly frozen by liquid nitrogen and sublimated in the vacuum desiccator. Thus, the freeze-dried samples retained the original microfabrics without any texture disruption and were best for SEM observation. The samples were coated by Au-Pd to observe the microfabrics by SEM at a setting of 15 kV and ~80 µA.
The embedded method for preparation of thin sections was conducted as follows. First, the pore water in the sediment was replaced by ethanol and then by propylene oxide for several months. Next, propylene oxide was further replaced by resin Quetol 651 from Nisshin EM Co. Ltd. Finally, the sediments were fixed under 60°C for 24 hr for thin sections.
