GEOCHEMISTRY
Chemical analyses of basalts, volcaniclastic rocks, and sedimentary rocks were determined during Leg 200 by ICP-AES. ICP-AES analysis proved difficult during the first half of the leg, mainly because of a problem in recording measurements using the software provided with the equipment. The dysfunction was evidently a flagging procedure in the data acquisition routine. Several protocols were investigated before the problem could be defined and corrected. Even when measurements were finally being recorded properly, however, neither of the two programs on board for calibrating to standards could be used successfully and we were compelled to evaluate the data largely step by step, using averaging and regression functions provided by standard application packages. This was fortunate, for we discovered some difficulties with particular elements that we would not have noticed if we had followed recommended procedures.
We selected representative samples of sedimentary rocks and volcaniclastic rocks (tuffs) from Hole 1223A and basalts from Site 1224 for shipboard ICP-AES analysis. Large whole-rock pieces were first cut with a diamond-impregnated saw blade and ground on a diamond wheel to remove surface contamination. Samples were washed in an ultrasonic bath containing methanol for ~10 min, followed by three consecutive ~10-min washes in an ultrasonic bath containing nanopure deionized water, and then dried for ~12 hr in an oven at 110°C. The cleaned whole-rock samples (~20 cm3) were reduced to fragments <1 cm in diameter by crushing between two disks of Delrin plastic in a hydraulic press and grinding for ~5 min in a Spex 8510 shatterbox with a tungsten carbide barrel. The sample powders were weighed on a Scientech balance and ignited to determine weight loss on ignition (LOI).
Aliquots of 100 ± 2 mg of the ignited whole-rock powders were weighed and mixed with 400 ± 0.4 mg of Li metaborate (LiBO2) flux that had been preweighed on shore. Standard rock powders and full procedural blanks were included with the unknowns for each sample run. All samples and standards were weighed on the Cahn Electro balance. Weighing errors are conservatively estimated to be ±0.01 mg.
Mixtures of flux and rock powders were fused in Pt-Au crucibles at 1050°C for 10-12 min in a Bead Sampler NT-2100. A total of 10 µL of 0.172-mM aqueous lithium bromide (LiBr) solution was added to the mixture, before fusion, as an antiwetting agent to prevent the cooled bead from sticking to the crucible. Cooled beads were transferred to 125-mL polypropylene bottles and dissolved in 50 mL of 2.3-M HNO3 by shaking with a Burrell wrist action bottle-shaker for an hour. After digestion of the glass bead, all of the solution was filtered to 0.45 µm into a clean 60-mL widemouthed polypropylene bottle. Next, 2.5 mL of this solution was transferred to a plastic vial and diluted with 17.5 mL of 2.3-M HNO3 to bring the total volume to 20 mL. The solution-to-sample dilution factor for this procedure is ~4000. Dilutions were conducted using a Brinkmann Instruments dispensette.
Major (Si, Ti, Al, Fe, Mn, Mg, Ca, Na, K, and P) and trace (Zr, Y, Sr, Ba, Ni, Cr, Sc, V, Cu, and Zn) element concentrations of powder samples were determined with the JY2000 Ultrace ICP-AES. The JY2000 sequentially measures characteristic emission intensities (with wavelengths between ~100 and 800 nm). Murray et al. (2000) developed protocols for dissolution and ICP-AES analysis of rock powders (see also Shipboard Scientific Party, 2001). The hard rock analytical procedure was refined during Leg 197 (Shipboard Scientific Party, 2002). The elements analyzed, the emission lines used, and the specific analytical conditions for each sample run during Leg 200 are provided in Table T1.
The JY2000 plasma was ignited 30 min before each run to allow the instrument to warm up and stabilize. After the warm-up period, a zero-order search was performed to check the mechanical zero of the diffraction grating. After the zero-order search, the mechanical step positions of emission lines were tuned by automatically searching with a 0.002-nm window across each emission peak using the University of Massachusetts Kilauea basalt laboratory standard K-1919, prepared in 2.3-M HNO3. The only exception is P, which was automatically searched by using a single-element standard. During the initial setup, an emission profile was collected for each peak, using K-1919, to determine peak-to-background intensities and to set the locations of background points for each element. The JY2000 software uses these background locations to calculate the net intensity for each emission line. The photomultiplier voltage was optimized by automatically adjusting the gain for each element using the standard (BHVO-2 or BIR-1) with the highest concentration for that element. Before each run, a profile of K-1919 was collected to assess the performance of the machine from day to day.
All ICP-AES data presented in the site chapter reports were acquired using the Gaussian analytical mode of the Windows 5 JY2000 software. This mode fits a Gaussian curve to a variable number of points across a peak and then integrates to determine the area under the curve. The parameters for each run are given in Table T1. Each unknown sample was run at least twice, nonsequentially, in all sample runs.
A typical ICP-AES run included (1) a set of three certified rock standards (BHVO-2, BIR-1, and BCR-2) (Table T2) run at the beginning, middle, and end of the sample run; (2) as many as 11 unknown samples; (3) a drift-correcting sample (the K-1919 standard) analyzed every fifth sample position, and at the beginning and end of each run; and (4) a blank solution run near the beginning, middle, and end of each run. A 2.3-M HNO3 wash solution was run for a minimum of 90 s between each of the samples and standards.
Following each sample run, the raw intensities were transferred to an Excel data file, and data reduction was completed using Kaleidagraph. The enhanced plotting functions in Kaleidagraph allowed quick assessment of drift and the repeatability of standards and the blank and identification of occasional discordant measurements. This ensured proper control over standardization and drift correction. Once transferred, a drift correction was then applied to each element and to the blank by linear interpolation between drift-monitoring solutions run before and after a particular batch of samples, assuming that the time between measurements during autosampling was constant. The interpolation was calculated using the lever rule. The drift correction was applied to the blank after noticing for some elements a substantial systematic drift for the blank itself, consistent with the trend of the K-1919 drift monitor. The average of the three drift-corrected procedural blank measurements was then deducted from the drift-corrected intensities of all samples. Following drift correction and blank subtraction, concentrations for each sample were calculated from linear regressions using the average intensity per unit concentration for the three standards measured, each one being measured twice during the run. The blank was also included in the regression with both its intensity and concentration set at zero. This calibration differs from that of previous legs (e.g., Leg 197), in that several standards were used, rather than normalization to a single standard, BHVO-2. The regression technique gave good correlation coefficients (>0.999) for most oxides and trace elements. It also revealed either important discrepancies between standards or problems with sample preparation in the cases of Cu and Zn, which accordingly are not reported in the site chapters.
Estimates of accuracy and precision for major and trace element analyses were based on fits to the regressions for BHVO-2, BIR-1, and BCR-2, the results of which are presented in Table T2. In general, run-to-run relative error by ICP-AES was better than 2% for the major elements. Run-to-run relative error for trace elements was generally <5%. Exceptions typically occurred when the element in question was near the detection limit of the instrument (see Table T1 for instrument detection limits).
Secondary minerals (e.g., vein and void fillings) were identified by X-ray diffraction using a Philips PW1729 X-ray diffractometer. Selected samples were taken from the short sections cored at Sites 1223 and 1224. Most samples were taken from small spots in veins or cavity fillings to identify materials with distinctive visual characteristics (the white vug, the green, red, or white mineral lining the vein, etc.). A few were from bulk sediment. The samples were freeze-dried overnight, then crushed to fine grain size using a mortar and pestle, and the powders were mixed in water slurries on glass slides. Upon drying, these were ready for X-ray analysis. Ni-filtered CuK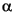 radiation was used. The instrument conditions were as follows: 40 kV, 35 mA; goniometer scan from 2° to 70°2
radiation was used. The instrument conditions were as follows: 40 kV, 35 mA; goniometer scan from 2° to 70°2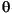 (air-dried samples); step size of 0.01°2; scan speed at 1.2°2/min; and count time of 0.5 s for each step. In most samples, carbonate minerals were not present, but where they were present we wished to determine their identity. Thus, no samples were decalcified. Also, because of the way the samples were selected, we did not attempt to separate or glycolate clays. MacDiff software (version 4.1.1 PPC, by Rainer Petschick) was used to display diffractograms; identifications are based on multiple peak matches, using the mineral database provided with MacDiff. Diffractograms were peak corrected to match the calcite peak at 3.035 Å. In the absence of calcite, no peak correction was applied.
(air-dried samples); step size of 0.01°2; scan speed at 1.2°2/min; and count time of 0.5 s for each step. In most samples, carbonate minerals were not present, but where they were present we wished to determine their identity. Thus, no samples were decalcified. Also, because of the way the samples were selected, we did not attempt to separate or glycolate clays. MacDiff software (version 4.1.1 PPC, by Rainer Petschick) was used to display diffractograms; identifications are based on multiple peak matches, using the mineral database provided with MacDiff. Diffractograms were peak corrected to match the calcite peak at 3.035 Å. In the absence of calcite, no peak correction was applied.
