BIOGEOCHEMISTRY
Interstitial Water Samples
Shipboard interstitial water (IW) samples were obtained from 5- to 30-cm-long whole-round intervals that were cut according to two general procedures. One set of IW intervals was cut on the catwalk, capped, and taken to the laboratory for immediate processing; the other set was cut from ends of shared microbiology cores that were subsampled in the walk-in refrigerator, usually within an hour of removal from the catwalk. During high-resolution sampling, when there were too many IW intervals to process immediately, the capped whole-round intervals were stored temporarily in a freezer or refrigerator. Cores with high contents of hydrogen sulfide were processed and stored temporarily in a fume hood.
After extrusion from the core liner, the surface of each whole-round interval was carefully scraped with a spatula to remove potential contamination. Sediments were then placed into a titanium squeezer, modified after the standard stainless steel squeezer of Manheim and Sayles (1974). The piston was positioned on top of the squeezer, which was then flushed with nitrogen through the outlet for >2 min. Pressures of up to 76 MPa were applied in the squeezer, calculated based on the measured hydraulic press pressure and the ratio of the piston areas of the hydraulic press and the squeezer. Interstitial water was passed through a prewashed Whatman number 1 filter fitted above a titanium screen, filtered through a 0.45-µm Gelman polysulfone disposable filter, and subsequently extruded into a precleaned (10% HCl) 50-mL plastic syringe attached to the bottom of the squeezer assembly. After collection of interstitial water, the syringe was removed to dispense aliquots for shipboard and shore-based analyses.
A modification of the above procedure was implemented to prevent loss of ephemeral constituents because of the backlog of interstitial water samples awaiting dispensing from the 50-mL syringes. In addition to the 50-mL syringe, IW samples were also collected in a 10-mL glass syringe attached to the squeezer assembly and the 50-mL syringe via a three-way plastic valve. Interstitial water emerging from the disposable filter at the bottom of the squeezer assembly was directed into one or the other syringe as necessary for appropriate dispensing of aliquots. Use of the 10-mL syringe also avoided air bubbles and minimized contamination of this fraction of the interstitial water by dissolved O2.
Interstitial Water Analyses
Most IW samples were analyzed for routine shipboard measurements according to standard procedures (Gieskes et al., 1991). Salinity was measured as total dissolved solids using a Goldberg optical handheld refractometer. The pH was determined by ion-selective electrode. Alkalinity was determined by Gran titration with a Metrohm autotitrator.
A new procedure was implemented during Leg 201 to analyze for dissolved inorganic carbon, employing a Coulometrics 5011 CO2 coulometer. An aliquot of 1.0 mL of interstitial water was pipetted into the reaction tube, followed by addition of 3.0 mL of 2-N HCl after attaching the reaction tube to the coulometer apparatus. The liberated CO2 was titrated, and the end point was determined by a photodetector. Measured concentrations were corrected for the value of the acid blank. Analytical uncertainty, based on repeated measurements of a sample of surface seawater, was ±1%.
Concentrations of chloride and sulfate were determined by manual dilution and manual injection into a Dionex DX-120 ion chromatograph. Chloride concentrations were also determined at some sites by titration with AgNO3. Quantification was based on comparison with International Association of the Physical Sciences of the Ocean (IAPSO) standard seawater.
Dissolved silica, phosphate, and ammonium concentrations were determined by spectrophotometric methods using a Shimadzu UV Mini 1240 spectrophotometer. For phosphate analyses at Sites 1227-1229, the standard protocol was slightly modified. Previous analyses of dissolved phosphate in interstitial waters of shallow Peru Margin sediments at Site 684 and also Sites 680 and 681 were strongly affected by color interference because of high concentrations of hydrogen sulfide (Shipboard Scientific Party, 1988). At the Peru Margin Sites 1227, 1228, and 1229, we aimed to improve upon the standard ODP technique for dissolved phosphate analysis in H2S-rich IW samples. One approach was to remove sulfide from the sample by acidification and degassing. Samples that had been titrated for alkalinity were used, as they are acidified and degassed. Furthermore, they are in a pH range appropriate for colorimetric determination of phosphate by the phosphomolybdate blue method.
Selected trace metal concentrations were obtained using the Jobin-Yvon Ultrace inductively coupled plasma-atomic emission spectrometer (ICP-AES). Concentrations of boron, barium, iron, lithium, manganese, and strontium were determined following the procedures outlined by Murray et al. (2000). Given the anticipated range of redox environments at Leg 201 sites and the fact that many microbially mediated reactions depend on metal catalysts, IW samples were also examined for a suite of redox-sensitive transition metals—copper, molybdenum, nickel, vanadium, and zinc. For these analyses, the shipboard "master" ICP-AES standard was modified so that concentrations of iron, manganese, lithium, boron, and strontium remained the same but with concentrations of copper, molybdenum, nickel, vanadium, and zinc at 200, 200, 200, 200, and 500 mM, respectively. Analytical standards for all elements were then prepared by analyzing mixtures of this master standard and seawater.
Dissolved sulfide (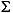 H2S = H2S + HS-) was determined on 1-mL interstitial water samples injected into a pre-tared vial containing 0.5 mL of 20% zinc acetate solution (20 g ZnAc/100 mL solution). Dissolved sulfide was determined by the methylene blue method of Cline (1969) using a Shimadzu UV Mini 1240 spectrophotometer and a Milton Roy Mr. Sipper sample introduction system. Iodometrically calibrated zinc sulfide suspensions in zinc acetate solution were used to calibrate the diamine reagent.
H2S = H2S + HS-) was determined on 1-mL interstitial water samples injected into a pre-tared vial containing 0.5 mL of 20% zinc acetate solution (20 g ZnAc/100 mL solution). Dissolved sulfide was determined by the methylene blue method of Cline (1969) using a Shimadzu UV Mini 1240 spectrophotometer and a Milton Roy Mr. Sipper sample introduction system. Iodometrically calibrated zinc sulfide suspensions in zinc acetate solution were used to calibrate the diamine reagent.
Nitrate and nitrite concentrations were determined spectrophotometrically on 1-mL samples according to the methods of Strickland and Parsons (1972) using a Shimadzu UV Mini 1240 spectrophotometer with a Milton Roy Mr. Sipper sample introduction system.
Volatile fatty acids (i.e., acetate and formate) were analyzed by ion exchange chromatography on a Dionex ion chromatograph equipped with an anion exchange column (Dionex AS-15). Solutions of sodium hydroxide and sulfuric acid were utilized as eluent and suppressant, respectively. A 1.0-mL sample of filtered interstitial water was slowly applied to a sequence of ion exchange cartridges to remove interfering ions (cartridge A: Dionex OnGuard II H packed with a Ag-form cationic resin to remove chloride followed by cartridge B: Dionex OnGuard II H packed with a proton-form cationic resin to remove excess Ag eluting from previous cartridge) and after a period of at least 5 min eluted with 1.0 mL H2O. Prior to use, cartridges were conditioned by rigorous flushing with deionized water (at least 20 mL). The detection limit for acetate was constrained by interferences with variable amounts of lactate and typically ranged from 0.2 to 0.5 µM. The detection limit for formate was 0.1 µM.
Hydrogen concentrations were determined on incubated sediment samples following published procedures that assume the headspace hydrogen is in equilibrium with the dissolved pore fluid hydrogen (Lovley and Goodwin, 1987; Hoehler et al., 1998). Four replicate incubations were conducted on each sample. For each incubation, a 5-cm3 bulk sediment sample was collected from a freshly exposed end of a core section using a sterilized plastic syringe with its Luer tip cut off. The sediment sample was extruded into a 20-mL headspace vial and immediately capped with a rubber septum that was sealed with an aluminum crimp cap. The sealed vials were flushed with low-hydrogen nitrogen using two hypodermic needles inserted through the septum. One needle was connected to the nitrogen and one allowed for gas release. These septa and vials were previously shown not to leak significant hydrogen over the timescale of the incubations. Headspace hydrogen concentrations were analyzed daily until approximately steady-state concentrations were reached. Hydrogen concentrations were determined by gas chromatography on a Trace Analytical reduction gas analyzer. Quantification was by comparison to a standard curve generated from a single primary gas standard and mixed to different concentrations immediately prior to analysis. To correct for drift, the primary gas standard was repeatedly analyzed. Reported concentrations are based on the temperature-dependent solubility of hydrogen and the mean of replicates.
Concentrations of methane were monitored at intervals of 2 to 17 samples per core. The standard gas analysis program for safety and pollution prevention purposes (Kvenvolden and McDonald, 1986) was complemented by additional headspace analyses following a slightly different approach (Iversen and Jørgensen, 1985; Hoehler et al., 2000) with the intent to better constrain the concentrations of dissolved gases. Compared to the rapid safety-oriented protocol, the latter, more time-consuming alternative led to higher yields of methane (e.g., see "Biogeochemistry" in the "Site 1225" chapter).
Upon core retrieval, a 3-mL sediment sample was collected with a cut-off plastic syringe from a freshly exposed end of a core section and was extruded into a 20-mL glass serum vial. For this purpose, the plunger was held at the sediment surface while inserting the barrel to avoid trapping air bubbles. After withdrawing the syringe, the plunger was advanced slightly to extrude a small amount of sediment. This excess was shaved off with a flat spatula flush with the end of the syringe barrel to provide an accurate determination of the sediment volume within the syringe. Samples required for safety and pollution prevention purposes were immediately sealed with a septum and metal crimp cap and heated to 60°C for 20 min. The headspace was subsequently analyzed by gas chromatography. For samples designated for refined headspace analysis, the sediment was extruded into a 20-mL vial containing 5 mL of 1-M NaOH. The vial was immediately capped with a silicone/Teflon septum. After vigorous manual shaking for 2 min, the vials were shaken automatically for an additional hour and subsequently left to stand for at least 23 hr at room temperature prior to gas chromatographic analysis. Additionally, when gas pockets were observed, headspace samples were complemented by vacutainer samples, which were collected directly from gas voids formed in the core liner by penetrating the liner using a syringe connected to a penetration tool.
Gas chromatographic analyses of headspace samples resulting from both protocols were performed in an identical manner. A 5-mL volume of headspace gas was extracted from the vial using a standard gas syringe. This volume was compressed in the syringe to a volume of 3 mL. The created overpressure was released by briefly opening the valve of the gas-tight syringe. Constituents of the headspace and vacutainer gas samples were analyzed using a Hewlett Packard 6890 Plus gas chromatograph (GC) equipped with an 8-ft x 1/8-in stainless steel column packed with HaySep S (100-120 mesh) and a flame ionization detector (FID). Concentrations of methane, ethane, ethene, propane, and propene were obtained. The carrier gas was helium, and the GC oven was programmed from 100°C (5-min hold) to 140°C (4.5-min hold) at a rate of 50°C/min. Data were collected using a Hewlett-Packard 3365 ChemStation data processing program. Gas samples collected with vacutainers were routinely analyzed on the natural gas analyzer (NGA). The NGA system consists of a Hewlett-Packard 6890 Plus GC equipped with three different columns and two detectors. Hydrocarbons from methane to hexane were analyzed using a 60-m x 0.32-mm DB-1 capillary column connected to a FID. The GC oven was heated isothermally at 50°C for 15 min.
The concentration of methane in interstitial water was derived from the headspace concentration by the following equation:
-
CH4 =
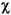 M · Patm · VH · R-1 · T-1 ·
M · Patm · VH · R-1 · T-1 · 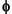 -1 · VS-1,
(1)
-1 · VS-1,
(1)
where,
-
VH = volume of the sample vial headspace,
-
VS = volume of the whole sediment sample,
-
M = molar fraction of methane in the headspace gas (obtained from GC analysis),
-
Patm = pressure in the vial headspace (assumed to be the measured atmospheric pressure when the vials were sealed),
-
R = the universal gas constant,
-
T = temperature of the vial headspace in degrees Kelvin, and
-
= sediment porosity (determined either from moisture and density measurements on adjacent samples or from porosity estimates derived from gamma ray attenuation [GRA] data representative of the sampled interval).
Quantities of methane that remain undetected because of dissolution in the aqueous phase are minimal (e.g., Duan et al., 1992) and are not accounted for. The internal volumes of 15 representative headspace vials were carefully measured beforehand and were determined to average 18.42 ± 0.13 mL. This volume was taken as constant in calculations of gas concentrations.
During Leg 201, we discovered that at most sites the concentrations obtained from the safety-related headspace protocol were significantly lower than those obtained from comparable samples analyzed by the prolonged extraction method using sediment slurries in NaOH solution. We suggest that the prolonged extraction solution led to the detection of a methane fraction that is not dissolved in interstitial water. For Sites 1229-1231, where particularly long extraction times had been applied and led to unusually high-yield increases, we consider the data obtained from the safety protocol as the best approximation for the fraction of dissolved methane. Future research will verify the nature of the additional pool of methane tapped by prolonged extraction in alkaline solution.
Sediment samples were not routinely analyzed during Leg 201 because of the emphasis on interstitial water and gas analyses and because all of the sites drilled during this leg were sampled during previous legs. However, some analyses were obtained on sediment samples according to the standard methodology employed during previous legs. Inorganic carbon (IC) concentration was determined using a Coulometrics 5011 CO2 coulometer. About 10 to 15 mg of freeze-dried, ground sediment was weighed and reacted with 2-N HCl. The liberated CO2 was titrated, and the end point was determined by a photodetector. Calcium carbonate, expressed as weight percent, was calculated from the IC content, assuming that all evolved CO2 was derived from dissolution of CaCO3, by the following equation:
-
CaCO3 (wt%) = 8.33 x IC (wt%).
(2)
No correction was made for the presence of other carbonate minerals.
Total carbon (TC), nitrogen, and sulfur concentrations were determined using a Carlo Erba 1500 CNS elemental analyzer. About 10 mg of freeze-dried, ground sediment was weighed and combusted at 1000°C in a stream of oxygen. Nitrogen oxides were reduced to nitrogen, and the mixture of carbon dioxide, nitrogen, and sulfur dioxide was separated by GC and detected by thermal conductivity detector (TCD). Total organic carbon (TOC) concentration was calculated as the difference between TC and IC concentrations.
The organic matter in selected sediment samples was characterized by pyrolysis using a Delsi Nermag Rock-Eval II system. This method is based on a whole-rock pyrolysis technique designed to characterize the type and maturity of organic matter and to estimate the petroleum potential of the sediments (Espitalié et al., 1986). The Rock-Eval system incorporates a temperature program that initially expels volatile hydrocarbons (S1) as the sample is heated at 300°C for 3 min and then, as the temperature increases from 300° to 600°C at 25°C/min, releases the hydrocarbons (S2) resulting from thermal cracking of kerogen. S1 and S2 hydrocarbons are measured and reported in milligrams per gram of dry sediment. The temperature at which the kerogen yields the maximum amount of hydrocarbons during the S2 program provides Tmax, a parameter used to assess the maturity of the organic matter. Between 300° and 390°C of the pyrolysis program, carbon dioxide (S3) is released from the organic matter, trapped, measured by TCD, and reported in milligrams per gram of dry sediment. Rock-Eval II parameters are used to characterize organic matter by calculation of hydrogen index (HI), oxygen index (OI), and S2/S3:
-
HI = (S2/TOC) x 100 and
(3)
-
OI = (S3/TOC) x 100.
(4)
Rock-Eval data are generally unreliable for samples containing <0.5 wt% TOC.
