 for precipitation of apatite) or no fractionation of oxygen in phosphate.
for precipitation of apatite) or no fractionation of oxygen in phosphate.
ODP's recent progress in exploring the deep subseafloor biosphere has revealed that prokaryotes are consistently present in core samples recovered from the deep oceanic subsurface (Parkes et al., 1994; Wellsbury et al., 1997). The subseafloor biosphere has been estimated to constitute one-third of the biomass on Earth (Whitman et al., 1998). However, the structure, diversity, and function of subsurface microbial communities remain poorly understood. Total cell numbers alone do not provide information about prokaryotic physiologies that are critical to understanding deep biosphere biogeochemical processes. It is important to know (1) what types of prokaryotes are present and in what abundance and (2) which of these prokaroytes are truly active (i.e., not dormant) and are participating in deep sedimentary geochemical processes. We used a range of approaches to quantify prokaryote abundance, diversity, and activity, including (1) total counts; (2) adenosine triphosphate (ATP); (3) cultivation methods, particularly the most probable number (MPN) technique; (4) nucleic acid-based techniques, particularly fluorescence in situ hybridization (FISH); and (5) radiotracer and stable isotope tracer experiments on specific microbial processes. In order to ensure that we were indeed analyzing the indigenous prokaryotes and their activities, tests for contamination were conducted during the entire coring process for microbiological samples.
Microbiological sampling depends on careful and appropriate sample handling technique. Precise operational definitions for special microbiology handling terminology is given in Table T1. Because the samples were retrieved from very stable sedimentary environments, the prokaryotes are expected to be sensitive to chemical and physical change, in particular to changes in oxygen, temperature, and (for the deep-sea sites) pressure. Consequently, all samples for microbiology and process studies were transferred from the drilling platform to the hold refrigerator (set to <10°C) as quickly as possible and were kept as whole-core sections until processed (to date, there is no system for retrieving and maintaining samples under in situ pressure). In order to avoid intermittent warming of retrieved cores, ODP's usual core handling procedure was modified. Once a core was retrieved, it was immediately transferred to the catwalk for labeling and cutting before the next core barrel was deployed. When piston coring at 3800 m water depth, this prolonged the drilling operation by 33% per core (from 63 to 84 min) but was considered important to prevent damage to heat-sensitive microorganisms. Efforts were also made to obtain APC cores even when this led to an increase in core recovery times, as APC cores were generally much less disturbed than XCB cores.
While drilling cores for microbiology, the potential for contamination with bacteria from the surface is highly critical. Contamination tests were continuously conducted using solutes (perfluorocarbon tracer [PFT]) or bacterial-sized particles (fluorescent microspheres) to check for the potential intrusion of drill water from the periphery toward the center of cores and thus to confirm the suitability of the core material for microbiological research. We used the chemical and particle tracer techniques described in ODP Technical Note 28 (Smith et al., 2000a). Furthermore, the freshly collected cores were visually examined for possible cracks and other signs of disturbance by observation through the transparent core liner. Core sections observed to be disturbed before or after subsampling were not analyzed further. Such disturbance phenomena are critical to the integrity of the core material and therefore also to its usefulness for microbiological studies.
A limited number of microbiological and related biogeochemical samples were collected on the catwalk as soon as the core was retrieved. After the core was cleaned and the IR camera scan completed, the core was marked into 1.5-m sections for cutting and visually inspected for signs of disturbance, such as gas voids, cracks, and drilling disturbance. The appropriate sections, usually from the middle, were taken for microbiological analyses. The top end of the selected sections were cut and capped (without acetone). The top 15 cm of this section (or the bottom 15 cm of the previous section) was often used as an interstitial water biogeochemistry sample. The lower end was cut, and a temperature image was immediately taken by a calibrated IR-sensitive video camera (see "Infrared Thermal Imaging" in "Physical Properties") to estimate the maximum temperature reached in the core center before it was transferred to the cold room. Samples for total prokaryotic cell counts and perfluorocarbon contamination checks were immediately collected using 5-cm3 sterile syringes from the lower, freshly cut end, whereas samples for methane and porosity were taken from the adjacent core section end. This catwalk syringe sampling enabled collection of these samples at a much greater frequency than was possible for the whole-round cores (WRCs). The lower core end was then sealed with an end cap (without acetone). The microbiological section and an adjacent section were quickly transported to the cold room to limit temperature increase (see "Infrared Scanner" in "Physical Properties" in the "Site 1225" chapter).
It is important to emphasize that the different analyses, experiments, and cultivation attempts that fall under the rubric "microbiological methods" have widely different requirements concerning handling and storage. Keeping samples cool, processing times short, and minimizing contamination were the key criteria for determining how the core sections were processed. To minimize changes in the microbial population, all handling took place in a cold room. The lower refrigerated core room on the hold deck of the ship served as a cold room at <10°C and was equipped with a work bench and working space for two to four persons. It was important that all materials, including core cutters, glass vials, and so on, were kept cold so that no unintentional warming of the samples took place. In addition, as the core liner is not sterile and the outer surface of the core is contaminated during drilling (Smith et al., 2000a, 2000b), subsampling must exclude the sediment next to the core liner. Where appropriate, handling and subsampling were performed under anoxic conditions.
Normally, two 1.5-m core sections at a time were brought down to the cold room in case the standard section or a part of this section was found to be disturbed on subsampling. In such cases, part or all of the subsequent section was also subsampled. Table T2 and Figure F6 show the various categories of samples and how they were handled. The subsectioning equipment included the standard ODP core cutter coupled with a clean wire or blade and a nitrogen-flushed cutting rig that was modified from an earlier published version (Fig. F7) (Parkes et al., 1995). The cutting rig system enabled a 1.5-m section, after cleaning the outer surface with ethanol, to be sequentially cut into a number of WRC sections using a sterile blade. Some of these WRC sections were immediately capped with clean end caps and stored at 4°C for shore-based analysis, whereas others were immediately subsampled into sterile 5-cm3 or larger syringes (with the Luer end removed).
To minimize contamination and to increase handling efficiency, several modifications of the core cutting procedure were introduced for microbiology subsamples intended for cultivation, starting from Site 1226. These include the following:
After Site 1225, the nitrogen-flushed cutting rig was used principally for samples requiring anoxic sampling conditions (e.g., activity measurements).
Anoxic subsampling with cut-off syringes was conducted under a flow of filter-sterilized nitrogen in a gassing "bucket" designed at Bristol University (Fig. F8). The bucket system prevents the nitrogen flow from creating turbulence and thus introducing contamination or oxygen. Syringe subcores were taken from the central uncontaminated part of the WRC and then sealed under nitrogen with a sterile stopper. To preserve the integrity of the sample during subcoring and to prevent sediment near the core liner from being sampled, sterile acrylic pegs were inserted into the holes left after each subcore was removed. Subcores were stored at 10°C under nitrogen atmosphere in gas-tight bags until further processing. In compacted sediments, it was occasionally necessary to drive the syringe into the WRC with a syringe adapter and hammer. Syringe subcores were used for various analyses, including radiotracer studies, production of sediment slurries for bacterial enrichments, MPN counts, FISH, bead contamination tests, and measurements of hydrogen concentration.
WRCs to be immediately preserved were sectioned with a standard ODP cutter and sterile wire from the same core. The samples for deoxyribonucleic acid (DNA) and lipid biomarker analysis were frozen in a -80°C freezer. Samples for iron, manganese, and sulfur solid-phase speciation and isotopes were placed in aluminum gas-tight bags, vacuum sealed, and frozen at -20°C. WRCs for further shore-based microbiological experimentation were stored in a nitrogen gas-flushed aluminum bag, often together with a welled Merck Anaerocult strip, and stored at +4°C. Clean disposable gloves were worn during all handling procedures. Any remaining portions of the sections used for microbiology subsampling were returned to the core laboratory for reintroduction into the standard core handling process. In order to obtain uncontaminated material for slurry preparation and cultivation, cores were broken after precutting the core liner with the ODP cutter. Bending the outer ends upward allowed released particles to drop into a bin. This technique provided untouched (although not always smooth) surfaces that were sampled by a 60-mL syringe. Only deeper sediment contents of the syringe, which did not contain oxygen, were transferred to nitrogen-flushed sterile slurry vessels containing artificial seawater. An overview of the cutting and subsampling scheme for the microbiology section is given in Figure F6 and in Table T2.
The most immediate method to visualize and quantify the deep biosphere are total prokaryotic cell counts using the nucleic acid stain acridine orange. These counts have been made on a wide range of ODP sediment cores, including cores from the Peru margin and the equatorial Pacific (Parkes et al., 1994). In general, these counts have demonstrated an exponential decrease of prokaryotic cells with depth. Prokaryotic cells were consistently detected, even in the deepest sediments. The method detects sediment layers of increased cell density that often coincide with particular geochemical conditions that are conducive to prokaryote growth (Parkes et al., 2000). The acridine orange direct count (AODC) enumeration method was used at all sites during this leg. Contamination during drilling and handling was evaluated by tests using micrometer-sized fluorescent beads and PFT. These tests have shown that core samples can be obtained without introducing prokaryotic cell contamination, which is essential for almost all microbiological analyses that follow core retrieval (Smith et al., 2000a, 2000b).
WRCs were cut in the cold room and stored at -80°C for shore-based analysis of ATP concentrations using the luciferin-luciferase assay. Adenosine-5´-triphosphate is used as a common currency of energy for all organisms on Earth. ATP is generated by energy-yielding reactions and is subsequently consumed in energy-requiring reactions in the cell. Because ATP molecules degrade rapidly upon cell death, ATP concentrations can be used as an indicator of total living biomass (Levin et al., 1964). This approach has been used in various marine environments (Holm-Hanson and Booth, 1966), including sediments (Karl and LaRock, 1975; Stoeck and Duineveld, 2000). ATP will be extracted from sediments and quantified using the luciferin-luciferase assay. These data will be compared to total cell counts in order to estimate the fraction of the observed community that is viable.
Using classic cultivation techniques such as the MPN cultivation method, various physiological types of prokaryotes have been enriched from deep sediments and their abundances determined (e.g., Parkes et al., 2000). The MPN method allows quantification of the number of viable prokaryotes according to a statistical evaluation of the number of tubes of different tenfold dilutions in which growth has been detected (Garthright, 2001). The prokaryotic types that have been cultured from sediment ODP obtained using the MPN method include aerobic ammonifiers, nitrate reducers, fermentative anaerobic heterotrophs, sulfate reducers, methanogens, acetogens, and anaerobic hexadecane oxidizers (Cragg et al., 1990, 1996; Bale et al., 1997; Barnes et al., 1998; Parkes et al., 2000; Wellsbury et al., 2000). MPN population counts range from 0 to 105 cells/cm3 and generally decrease with increasing depth. By MPN enumeration, however, generally far fewer than 0.6% of the total cell numbers in deep sediments are detected, and these viable counts thus yield only limited quantitative information about the microbiology of the deep subsurface. In surface sediments, higher MPN counts have been obtained by the use of complex low-substrate media prepared from sediment extracts and containing fine particles (Vester and Ingvorsen, 1998). Such an approach was used during Leg 201 for the first time with deep sediments.
Cultivation is the only way to obtain microorganisms and study their physiology in order to estimate their impact on biogeochemical cycles in deep sediments. For this reason, we enriched (with the aim to isolate and characterize) various types of microorganisms using a wide range of media and culture conditions that covered a wide range of environmental conditions and metabolic requirements. Without going into detail about the media, which are listed in "Enrichments near In Situ Temperatures" in "Methods for Enrichment and MPN" in "Procedures and Protocols," the following groups of prokaryotes were targeted:
Culture-independent molecular ecological surveys are becoming an indispensable and powerful approach to investigating naturally occurring microbial diversity. Molecular community analysis of deep subsurface microbial ecosystems also offers new ways to understand the relationship between microbial community composition, microbial activities, and the biogeochemical characteristics of the sedimentary microbial biosphere (Sahm et al., 1999; Madson, 2000; Marchesi et al., 2001). Multiple molecular analytical techniques were applied to the deeply buried sediments drilled during Leg 201. These techniques are briefly described in the following subsections.
Molecular phylogenetic analyses are frequently based on the 16S ribosomal ribonucleic acid (rRNA) sequence. The 16S rRNA is an essential component of each ribosome, the multienzyme complex that translates messenger RNA into proteins, and therefore is a universal component of every living cell. Because of strict functional constraints, the 16S rRNA evolves very slowly and shows clearly recognizable homologies (similarity due to shared evolutionary ancestry) for all living organisms. In other words, the 16S gene is a short but central page from the book of life (~1500 letters) that has survived continued copying and editing for 3.5 billion years. The evolutionary division of life into the three domains of Bacteria, Archaea, and Eukarya is based on ribosomal RNA (Woese et al., 1990). In microbiology, 16S rRNA sequence analysis allowed for the first time a natural classification of microorganisms (including Ludwig and Schleifer, 1999). The analysis of 16S rRNA genes from mixed microbial communities in natural environments has, in combination with biomarker studies (see "Molecular Biomarkers"), opened a new way to determine microbial community structure without cultivation. By such techniques, numerous entirely new and so-far uncultivated phylogenetic lineages of prokaryotes have been discovered (Barns et al., 1996; Hugenholtz et al., 1998). In various microbial ecosystems, rRNA surveys have demonstrated that microbial diversity is much greater than previously assumed based on cultivation and isolation methods. During the past decade, extensive surveys of extreme environments such as geothermal hot springs, deep-sea sediments, or hydrothermal vent fields (Takai and Horikoshi, 1999; Li et al., 1999; Inagaki et al., 2001; Takai et al., 2001; Inagaki et al., in press; Teske et al., 2002) using culture-independent molecular ecological techniques have extended our knowledge of the phylogenetic diversity in naturally occurring microbial communities.
Many prokaryotes have unique metabolic and biochemical properties that are not found elsewhere in the living world. For example, anaerobic respiration with sulfate as an electron acceptor (sulfate reduction), carbon dioxide reduction to methane (methanogenesis), and acetate synthesis from carbon dioxide and hydrogen (acetogenesis) are exclusively prokaryotic processes. The specific enzymes that catalyze these processes are coded by key genes that are unique for these metabolic pathways. Thus, the presence of a key gene in a prokaryote is indicative of the corresponding key enzyme and the metabolic capacity of this organism. Because of functional constraints (the amino acid sequence has to remain conserved to ensure proper enzyme function), many of these key enzymes and their genes are highly conserved in their protein and nucleic acid sequence. A comparison of different versions of the same key gene from different prokaryotes reveals the following:
In addition to the well-documented 16S rRNA gene (~10,000 sequences in the databases and rapidly growing) (Maidak et al., 2001), databases and assays for several phylogenetically informative and functionally conserved metabolic key genes have been developed. Some key genes that will be studied postcruise in samples from Leg 201 and are specifically relevant for anaerobic subsurface prokaryotic populations and their activities are dissimilatory sulfite reductase (dsrAB) and adenosine-5´-phosphosulfate (APS) reductase for bacterial sulfate reduction (Wagner et al., 1998; Klein et al., 2001; Perez-Jimenez et al., 2001), coenzyme-M methyl reductase for methanogenesis (Springer et al., 1995; Lueders et al., 2001), formyl tetrahydrofolate synthase for acetogenesis, benzoyl-coA reductase, and group I/II dehalogenases for degradation of complex organic compounds. This list can be easily extended. The diversity and occurrence patterns of these key genes will be correlated with biogeochemical measurements of the microbial processes and with microbial counts using general stains and fluorescent in situ hybridization (FISH) probes (see next section, FISH).
A powerful technique to quantify prokaryotic cells in environmental samples is FISH (Amann et al., 1990, 1995). In this approach, fluorescently labeled oligonucleotides are used to stain individual prokaryotic cells according to their phylogenetic affiliation. The probes hybridize with a universal phylogenetic marker molecule, the 16S ribosomal ribonucleic acid (16S rRNA), a polyribonucleotide of ~1500 bases that is an integral component of the ribosome, and therefore of every living cell (see "16S rRNA Gene"). The 16S rRNA contains phylogenetically informative sequence regions that range from universally conserved to genus and species specific (Ludwig and Schleifer, 1999) and present a large variety of target sites for FISH techniques of defined specificity. Because high rRNA content is indicative of actively metabolizing bacteria, FISH can provide quantitative information about active prokaryotes in an environmental sample. Using a fluorescence microscope, cells can be visualized and counted after fixing sample material and performing the hybridization. FISH has been successfully applied to quantify sulfate-reducing bacteria and other phylogenetic groups of prokaryotes in near-surface marine sediments (Llobet-Brossa et al., 1998; Boetius et al., 2000; Ravenschlag et al., 2000, 2001). An important characteristic of the technique is that a sufficient content of cellular ribosomes is a prerequisite for its successful application in sediments (Amann et al., 1995). To the date of Leg 201, the deepest positive FISH result is from <0.02 mbsf (Ravenschlag et al., 2001). Thus, the usefulness of FISH to quantify low-abundance and low-activity prokaryotes in deep sediments had to be evaluated during this cruise. Newer protocols for sediments have been published and were used for shipboard analyses (Pernthaler et al., 2001).
A novel approach, FISH-SIMS, combines FISH with secondary ion mass spectrometry (SIMS) and allows the analysis of stable carbon isotopic compositions of individual cells or cell clusters in environmental samples that are identified using nonspecific fluorescence stains or FISH probes (Orphan et al., 2001). This approach will be tested in shore-based research with deep subsurface samples.
Living bacterial and archael populations may be identified through structural analysis of prokaryotic polar lipids. Such molecular biomarker evidence can provide additional evidence as to the identity and abundance of various prokaryotic groups. Isotopic analysis (i.e., 13C) of intact polar lipids can provide information on metabolic pathways, in particular methylotrophy. These frozen (-80°C) samples will be analyzed on shore.
A range of radioisotope experiments were initiated on board the JOIDES Resolution during Leg 201. Because radioisotope studies had not previously been carried out on board the JOIDES Resolution, we include descriptions of the rationale, procedures, and safety protocols for radioisotope use in this chapter as a reference for future researchers. A radioisotope van equipped specifically for and dedicated to radioisotope experiments was purchased and outfitted by ODP/Texas A&M University (TAMU) prior to departure from San Diego. This van was installed above the Core Tech shop on the port side of the ship, aft of the rig floor. All radioisotope work was carried out in the restricted area in the isotope van. Every scientist who worked in the radioisotope facility (four scientists during Leg 201) was experienced in performing these radiotracer experiments and was required to provide documentation of such from his or her home institution. Only low-energy beta emitters were used during the cruise. Extensive care was taken to avoid any radioactive contamination that could be a potential problem for other research, particularly for the sensitive analyses of natural radioisotopes.
After subsampling according to the methods described in "Core Handling and Sampling" in "Introduction and Background," the subsamples dedicated to radioisotope studies were hand carried in a cool box and placed in the appropriate incubator in the van. Sample processing was conducted in a plastic tray on a laboratory bench covered with plastic-backed absorbent paper in the back 10°C room. All solutions were stored in tightly capped containers, and routine postwork contamination wipe tests were performed and logged. All usage of radioactivity was logged, and all contaminated laboratory products were stored in the van. Unused radioactive solutions and contaminated laboratory products remained in the van for transfer to TAMU for final disposal.
The use of radiotracers is critical to the analysis of prokaryotic activities in the deep subsurface. Such prokaryotic processes as sulfate reduction, methanogenesis, acetogenesis, and methane oxidation take place in deep sediments at extremely low rates, sub-nanomole per cubic centimeter per day or even sub-picomole per cubic centimeter per day (Wellsbury et al., 1997). Such rates are a hundredfold to a millionfold lower than process rates found in surface sediments of the continental shelf and upper slope (Jørgensen, 1982; Ferdelman et al., 1999; Fossing et al., 2000). Interstitial water gradients of sulfate, bicarbonate, or methane may be used in diffusion-diagenesis models to calculate the in situ rates of these processes. However, such models generally cannot take product recycling into account, and therefore they provide net transformation rates rather than gross rates. The difference between net and gross process rates cannot, for the most part, be estimated, but it may be large. As an alternative to modeling, the gross process rates may be determined experimentally in freshly retrieved ODP cores by incubating sediment samples in which the prokaryotes are still in their original physiological state.
Because of the high concentrations of substrates and products such as sulfate, bicarbonate, methane, and so on, it is not possible to detect the small concentration changes during incubations within a realistic time period. Long incubations (exceeding months to years) would be required, leading to strongly altered chemical conditions and prokaryote populations in the sediment samples. Therefore, the determined rates would no longer reflect in situ conditions. By the use of radiotracers, however, rate determinations in incubated sediment samples may become >10,000-fold more sensitive than when using chemical measurements alone (see "Sensitivity of Process Rate Determinations"). Only by radiotracer techniques is it possible to conduct direct experimental measurements of the most important microbial processes in the deep subsurface, and even then, such methods are operating close to their detection limits when studying million-year-old sediments. Consequently, the amounts of radioactivity applied and incubation times used must be higher than those normally used in studies of near-surface sediments.
It is critical to all experimental process measurements that the sediment remains as intact as possible. Otherwise, data become unreliable. Successful process studies require the following:
The following example demonstrates why the use of radioactive isotopes as tracers in experimental process studies may enhance the detectability by many orders of magnitude. The example illustrates a measurement of sulfate reduction in the middle of the sulfate zone of a sediment core. We assume that the interstitial water sulfate concentration is about one-third that of seawater (i.e., 10 mM). By normal ion chromatography, this concentration may be determined with an accuracy of ±1%. In order to detect sulfate reduction in an incubated sediment sample by chemical analysis alone, the sulfate concentration would thus need to change by at least 1% of 10 mM (i.e., by 0.1 mM or 10-4 M). If the reduction rate is, for example, 1 pmol/cm3/day = ~10-9 M/day, then the experiment must run for at least 105 days, which is 300 yr.
If, instead, radioactive sulfate is added to the sediment in trace amounts, the sulfate reducing bacteria will reduce this labeled sulfate at the same relative rate as they reduce the nonradioactive sulfate (i.e., the same fraction of radiolabeled and interstitial water sulfate will be reduced per unit time). With the use of radiotracer, the reduction of a much smaller fraction of the sulfate can be detected. This is not because the determination of sulfate radioactivity is substantially more accurate but because the radioactive sulfate is converted into radioactive sulfide that can be detected instead. After the experiment has been terminated, the sulfide is converted to hydrogen sulfide gas by acidification of the sediment, and this gas can be very efficiently separated from the sediment and from the radioactive sulfate. As the hydrogen sulfide starts out with a radioactivity of zero, it is possible to detect the reduction of <10-6 of the radioactive sulfate. A 10-6 fraction of 10 mM sulfate is 10-8 M sulfate (i.e., 10,000-fold lower than the minimum detectable fraction by chemical analysis). This would reduce the minimum duration of the experiment to 10 days, which is obviously more realistic for the length of an ODP cruise. This calculation demonstrates only the minimum experimental time required to detect sulfate reduction at a rate of 10-9 M/day. To obtain reasonable quantitative data for the process, the incubation time would need to be somewhat longer (e.g., 1 month), which is still practical. Similar calculations may be performed for 14C-labeled acetate, bicarbonate, and methane. In addition, the 35SO42- tracer will also be used in part of the MPN experiments for sensitive detection of active bacteria (cf. Vester and Ingvorsen, 1998) (see "Radiotracer MPN Experiments" in "MPN Analyses near In Situ Temperature" in "Procedures and Protocols").
Stable isotopes (e.g., 13C, 15N, and 34S) are important "natural" tracers of microbiological processes and are also useful in labeled-tracer studies, such as, the 13C-labeled substrates used in FISH-SIMS experiments described in a previous subsection. Although widely used in biochemical research but only rarely applied in biogeochemical/microbiological studies, oxygen isotopes are also powerful tracers of enzymatic/metabolic reactions. Varner and Kok (1967) first proposed the use of oxygen isotopes in water as a means for detection of enzymatic activity in extraterrestrial soil samples to be collected during the Mars Viking missions. The basis for using oxygen isotopes in water is that biologically active oxy-anions such as sulfate, nitrate, phosphate, or acetate may undergo rapid oxygen isotope exchange with water at low temperature, but only in reactions that are catalyzed by enzymes. Thus, the presence of enzyme catalytic activity, detected by the transfer of 18O from labeled oxy-anion substrates to water, implies the presence of life. Using this approach with 18O-labeled phosphate, Varner and Kok (1967) report a detection limit of 0.05% change, which during a 24-hr incubation period might be achieved by 104 active prokaryotic cells per gram sediment. A modified version of this approach involving 18O/16O ratios of phosphate and sulfate rather than of water was employed for Leg 201 sediment samples to assess the utility of stable oxygen isotope tracers in detecting metabolic activity in deep-sea sediments characterized by very low rates of microbial growth. This primarily shore-based research was initiated during Leg 201 to optimize the survival and growth of microbes in long-term 18O-label incubation experiments.
One advantage of using oxygen isotope ratios of phosphate and sulfate as reaction tracers is the ability to carry out incubation experiments over relatively long periods of time. Radioisotope tracers such as 35S are useful over weeks to a few months (half-life of 35S decay = ~3 months), whereas incubations that utilize changes in phosphate oxygen isotope ratios as a reaction tracer can be carried out over many months to years. This is because 18O is a stable isotope and P-O bonds are highly resistant to chemical cleavage at low temperature, resulting in extremely low rates of oxygen isotope exchange between phosphate and water, on the order of 105 yr at 25°C (Lécuyer et al., 1999). Phosphate solutions used as laboratory standards and stored at room temperature show no change in phosphate oxygen isotope composition (18OP) over several years. Further, abiotic/nonenzymatic reactions involving phosphate, such as mineral precipitation/dissolution, produce little (~1 for precipitation of apatite) or no fractionation of oxygen in phosphate.
Potentially contaminated sediment was removed with a sterile scalpel. A 1-cm3 minicore was then taken with a sterile 5-mL plastic syringe. The syringe was sealed with a sterile Suba-Seal stopper. In a clean area of the laboratory, the 1-cm3 plug was extruded into a sterile serum vial containing 9 mL of 2% (v/v) filter sterilized (0.2 µm) formaldehyde in 3.5% NaCl. The vial was crimped and shaken vigorously to disperse the sediment particles. Where cell counts were performed on sediment slurries, then a 2-mL volume of slurry was added to a sterile serum vial containing 9 mL of 2% (v/v) filtered sterilized (0.2 µM) formaldehyde in 3.5% NaCl.
Total prokaryotic cell numbers and numbers of dividing or divided cells were determined using acridine orange as a fluorochrome dye with epifluorescence microscopy (Fry, 1988). Fixed samples were mixed thoroughly, and a 5- to 50-µL subsample was added to 10 mL of 2% (v/v) filter-sterilized (0.1 µm) formaldehyde in 3.5% NaCl. Where sediments contained significant amounts of carbonate, then the 2% formaldehyde was made up in 3.5% NaCl and 2% acetic acid. Acetic acid dissolves a substantial amount of carbonate, allowing larger samples to be processed, and thus resulting in greater accuracy and lower detection limits. Acridine orange (50 µL of a 1-g/L filter-sterilized [0.1 µm] stock solution) was added, and the sample was incubated for 3 min. Stained cells and sediment were removed on a 0.2-µm black polycarbonate membrane (Osmonics, USA). Excess dye was flushed from the membrane by rinsing with a further 10-mL aliquot of 2% (v/v) filter sterilized formaldehyde in 3.5% NaCl, and the membrane was mounted for microscopic analysis in a minimum of paraffin oil under a coverslip.
Mounted membranes were viewed under incident illumination with a Zeiss Axiophot microscope fitted with a 100-W mercury vapor lamp, a wide-band interference filter set for blue excitation, a 100x (numerical aperture = 1.3) Plan Neofluar objective lens, and 10x oculars. Prokaryote-shaped fluorescing objects were enumerated, with the numbers of cells on particles doubled in the final calculation to account for masking by sediment grains.
The percentage of cells involved in division has been suggested as an indication of growth, although the assessment of dividing cells has never had a standardized approach in the literature. Dividing cells were defined operationally as those having clear invagination. A divided cell is operationally defined as a visually separated pair of cells of identical morphology. The percentage of cells involved in division is then calculated as follows:
The detection limit for prokaryotic cells is usually 1 x 105 cells/cm3 (Cragg, 1994); however, the use of acetic acid with carbonate sediments reduced this to 4.5 x 104 cells/cm3.
As commonly applied to subseafloor sediments, nucleic acid stains such as acridine orange only provide information on the presence or absence of cells in the sediment matrix. Such use does not demonstrate if these cells are alive and active, dormant, senescent, or even dead. A number of stains have recently been used to determine proportions of living and dead cells, predominantly in laboratory cultures. One stain that appears to have the potential for use with environmental samples is CFDA/SE (5[6]-carboxyfluorescein diacetate/succinimidyl ester; Molecular Probes, Oregon, USA). Samples were obtained from cores for shore-based analysis. The procedure requires a 48-hr incubation in phosphate-buffered mineral salt solution and observation under UV light (Fuller, et al., 2000).
PFT was continuously fed into the seawater drill fluid at a tracer concentration of 1 mg/L seawater drill fluid. Concentrations of PFT were measured in all sections used for microbiological studies. A 5-cm3 subcore from the adjacent section was routinely taken, as described by Smith et al. (2000a). In many cases, two 5-mL subcores were taken and placed in the same headspace vial to increase sensitivity. Further, a 5-mL sample was tested from the "master slurry" prepared in the cold room for microbial incubation experiments. Air samples were occasionally taken to monitor the ambient concentration of PFT in the cold room air or on the catwalk. The concentrations of PFT at the outer periphery of the drill cores were measured to verify delivery of the PFT. Additionally, during APC drilling at each site, the concentration of PFT was measured in radial transects from the edge of the core to the middle in an "X" pattern, providing a two-dimensional view of contamination from four separate edge points to the common center. Finally, during XCB drilling, chunks of intact core (biscuits) surrounded by slurries of sediment and drilling fluid were collected, measured in length, and digitally photographed. PFT concentrations in the slurry, at the edge of the intact core on fracture surfaces, and in the middle of the chunk were measured, thus providing data on the minimal size and quality of intact core pieces that could be confidently sampled for microbiological investigations.
The conditions used on the GC were somewhat different than those previously used by Smith et al. (2000a). We used a HP-PLOT/AL203 "S" deactivated column with film thickness = 50 µM, length = 15 m, phase ratio = 12, and column ID = 0.25 mm. The inlet temperature was 180°C with 10 psi, the detector temperature was 250°C, and the column temperature was 100°C for 3.5 min and then ramped up 50°C/min until it reached 200°C. The PFT peak was at a retention time of 5.1 min. We used a 1-mL injection. Larger injections were found to result in loss of material.
The routine that gave the best results was to first bake the sample headspace vial and the syringe at 80°C, then inject clean nitrogen gas onto the column to make sure that no PFT peak resulted from residual PFT in the syringe or in the GC. After a clean run was achieved, the sample was injected using the same baked syringe. At the time of the injection, the syringe was also still hot so PFT would not condense out before injection. Also, for best results, background air samples need to be taken regularly from the same location that is used for capping headspace vials (ideally on the catwalk when no core is present). Finally, cleaning PFT out of used syringes is critical. We found it was best to use a large Hamilton syringe (10 mL) that could be flushed several times with wash methanol to remove the PFT. The syringe had to be baked for a long time to remove the methanol in order to avoid having an interfering GC peak. It was also found that cores with high levels of sulfide resulted in GC traces with small air peaks, presumably from the sulfide scrubbing out oxygen in the headspace vial. Therefore, for cores rich in sulfide, the air peak cannot be used to normalize various GC runs. The PFT detection limit reported for Leg 201 sites was not set as a lower limit of the ability to detect PFT by the GC, but as a lower limit of ability to confidently assess the presence of PFT in real samples given the uncertainty inherent in subtracting background levels of PFT and the reliability of the integration of small GC peaks.
A suspension of submicron-sized fluorescent microspheres was introduced into the drill fluid at all stations at selected depths that were also used for microbiological sampling. A plastic bag with a suspension of beads, positioned within the core catcher, released the beads inside the core barrel as it hit the sediment, thus providing maximum effectiveness of the beads as tracers of potential bacterial contamination. The procedure for assessing this particle contamination was adapted from that used during Leg 190 (Smith et al., 2000a, 2000b). The sediment sample (5 mL of sediment or 10 mL of 25% sediment slurry) was mixed with an equal volume of saturated sodium chloride solution. The solution was centrifuged (Marathon 10K; 5 min; 2800 x g), and the supernatant was filtered onto black polycarbonate filters (0.2-µm pore size). Fluorescent microspheres were counted under UV light, and data are reported as number of microspheres per gram of sediment.
When comparing the results of both contamination tests, the presence of beads and PFT concentration inside 5-cm3 subcores, one should consider some important aspects. First, in contrast to the beads, PFT can travel through very small pore spaces and is found in the laboratory air and on the hands of anyone who has handled a core liner. Therefore, although its presence at high concentrations in a sediment sample (>0.1 ng PFT/g sediment) may suggest contamination, it is not necessarily an indication that microorganisms from the drilling fluid have in fact contaminated the sample. On the other hand, the absence of PFT from a sample indicates that contamination by drill water has not occurred.
Second, whereas the number of beads (5 x 1011 beads/20-mL bag) that are deployed is equivalent to the number of bacteria in ~1000 L of seawater (assuming 5 x 108 bacteria/L), their deployment does not produce a uniform dispersion along the core. Although PFT can always be found in sediment samples taken from the edge of cores, the same is not true for the beads. At this point, without knowing the factors that control the final concentration and distribution of beads along the core barrel, one should consider the beads as a qualitative rather than a quantitative measure of contamination. The presence of beads is a strong indication that contamination by prokaryote-sized particles from the drilling water has occurred; the absence of beads alone does not mean that a sample is uncontaminated.
Here we describe the subsampling from WRCs and the media, incubation conditions, and research rationales for the shipboard microbiologists who were using media for cultivation and quantification of subsurface prokaryotes. The different kinds of media were inoculated from a sediment slurry or directly from WRC material, both kept cold and anoxic. For quantitative enrichments using the MPN technique, parallel assays were inoculated from a dilution series (Fig. F9). For MPN evaluation and enrichments in general, the assays will continue to be incubated in different shore-based laboratories at different temperatures after the cruise, with the goal to quantify particular prokaryotic physiological types and to isolate pure cultures.
Slurries were prepared in 250-mL Buchner flasks closed with stoppers that allowed sterile nitrogen flushing by means of a gas filter and a stainless steel tube. The side arms could be closed with sterile stopcocks. A salt solution (150 mL, containing 23.5 g NaCl and 10.8 g MgCl2·6H2O per liter) and a magnetic stirring bar were added and autoclaved at 121°C for 20 min, then the solution was cooled under nitrogen. A slurry (25% v/v) was prepared by adding the sediment contents of 10 x 5-mL or 2 x 25-mL syringes under a nitrogen counterflow and sterile conditions. The slurry was homogenized by repeated shaking and vortexing at 0°C for 30 min. Subsamples of this master slurry were distributed to all research groups conducting total cell counts, contamination checks, cultivation (five groups), FISH analyses, 13C and 18O experiments, and measurement of hydrogen production.
To enrich prokaryotes, the different media, which are described in detail in "Enrichments near In Situ Temperatures" were inoculated with subsamples from the slurry or with intact sediment. To obtain slurry subsamples, sterile syringes were first flushed with nitrogen and then filled with the slurry via the Buchner flask side arm. As soon as possible, the media were inoculated, either in an anaerobic chamber or, in cases when Hungate or Balch tubes were used, directly on a laboratory bench (Balch et al., 1979; Widdel and Bak, 1992). For MPN counts, the subsamples were used to make a dilution series in tenfold dilution steps (Fig. F9). To each tube containing 9 mL of medium, 1 mL of inoculum from the previous dilution step was added under an nitrogen atmosphere. From each dilution tube, three (or up to six) tubes of an identical medium were inoculated in parallel.
The tubes were incubated on the ship and shipped back to home laboratories for further incubation at the specified temperatures for periods of several months to >1 yr. The long incubation periods are necessary to allow slow-growing prokaryotes time to develop a detectable population size in the culture tubes. For example, the first MPN counts of cold-adapted sulfate-reducing bacteria from Arctic surface sediments required 2 yr before full development (Knoblauch et al., 1999a). Subsamples will be taken postcruise from the highest dilutions showing positive growth and used for inoculation of new sterile dilution series. The highest dilutions are used for further transfer to maximize the probability of transferring and ultimately isolating dominant representatives of the microbial populations.
The in situ temperatures in the sediments drilled at Leg 201 sites ranged mostly from 1° to 10°C in the upper 100 m at the deep-sea sites and 12°-25°C at the continental shelf sites and in the lower part of some deep-sea sites (see "Physical Properties" sections in each site chapter). Accordingly, psychrophilic (cold-adapted, with temperature optimum <15°C) prokaryotes are most relevant for the ongoing biogeochemical processes in much of the deep sea, and mesophilic (temperature optimum between 20° and 45°C) prokaryotes are most relevant on the shelf.
This section describes the enrichment and MPN methods that were applied for fermenting prokaryotes, sulfate-reducing bacteria, and methanogens during Leg 201. Most of the following media were based on artificial seawater (marine salts medium [MM]) (see Table T3). After autoclaving and cooling under nitrogen, bicarbonate buffer, a vitamin mixture, and FeCl2 plus Na2S were added to this MM. Upon addition, the latter two compounds form black FeS precipitate at the bottom of the vial. The FeS functions as a reducing agent and microenvironment for prokaryotes. Sediment prokaryotes are not accustomed to living in a pure liquid environment and often attach to particles. In contrast to most classic media, those made from MM are thus turbid from the beginning and require a microscopic or molecular biological analysis to detect growth. Different carbon sources were added to the MM as specified in Table T4.
Monomer medium (Mono) (Table T4) contained 36 monomers, among them the 20 most common amino acids and many compounds that are intermediates in the anaerobic prokaryotic degradation of organic matter and are used by fermenting, sulfate-reducing, or methanogenic prokaryotes. In contrast to most classic media, the concentration of the substrates is low, as it is known that prokaroytes from low-substrate environments might be killed by high substrate concentrations.
Polymer medium (Poly) (Table T4) contained nonsoluble substrates that under anaerobic conditions require sequential cooperation of different prokaryotes for degradation. Chitin is present in many marine organisms. Xylane and cellulose are common polysaccharides. Peptone contains poly- and oligopeptides. It is expected that fermenters, sulfate-reducing bacteria, and methanogens will grow in this medium.
The medium with aromatic compounds and long-chain fatty acids (Aro) (Table T4) contained substrates that are usually only slowly degraded. Those substrates are typical for low-substrate environments, where easily degradable low molecular weight substrates have been depleted.
The lactate medium (Lac) (Table T4) used is a classic substrate for sulfate-reducing bacteria. It might be partially degraded to acetate by incompletely oxidizing sulfate reducers (Widdel and Bak, 1992) or fermented to propionate and acetate. Acetate can then be used by completely oxidizing sulfate reducers or methanogenic archea.
Sulfate-free medium (no-SO4) (Table T4) was used for sulfate-poor sediment layers in order to compare with sulfate-containing medium. This comparison should indicate how sulfate limitation influences the prokaryotic communities. Sulfate-free medium was prepared with MM salts to which Na2SO4, Na2S, and FeCl2 had not been added. In this medium, titanium(III)citrate (Zehnder and Wuhrmann, 1976) was used as the reducing agent.
Sediment extract medium (Sed) (Table T4) was used to offer a complex mixture of substrates containing many natural substrates. It does not reflect the natural situation, since the extracted sediment was rather young and was treated with heat. However, it has been found that such media may support the growth of more types of prokaroytes than those composed of defined compounds. The extraction solution had the same composition as the MM salts, except that it did not yet contain NaHCO3, vitamins, Na2S, and FeCl2. 4-(2-Hydroxyethyl)piperazine-1-ethansulfonic acid (HEPES) was added as a buffer. Sediment from the 5- to 20-cm subsurface layer of North Sea tidal flats was mixed with the same weight of extraction solution and stirred overnight at room temperature. The mixture was then heated to 80°C for 30 min. After cooling and sedimentation of larger particles, the supernatant was filtered through a cellulose filter. The extract was autoclaved and cooled under nitrogen before vitamins, FeCl2, and Na2S were added to the same concentration as to the MM salts.
MPN counts of psychrophilic and mesophilic microorganisms were performed using deep-well titer plates (8 wells x 12 wells; Beckmann 27007) at temperatures of 4° or 15°C (Fig. F10). Wells were filled with 900 µL of medium under nitrogen. Then, 100 µL of master slurry was added, and triplicate tenfold dilution series were made down to 10-6. For each triplicate dilution series, a row remained uninoculated as a control. Plates were closed by sterile capmats (Beckmann 267005) and stored in anoxic bags under nitrogen with oxygen scrubber trays (Anaerocult, Merck). The media turbidity (because of iron sulfide precipitation) and the limited growth (because of low substrate concentrations) require the wells to be analyzed by microscopy, fluorescence microscopy, molecular biology, or with radiotracers (see "Radiotracer MPN Experiments"). The incubation time at 4°or 15°C will be up to 2 to 6 months.
Under anoxic conditions, many substrates are degraded syntrophically by cooperation of different groups of bacteria. Fermenters require the activity of hydrogen-consuming sulfate reducers or methanogens. In order to promote syntrophic growth, sulfate-reducing bacteria were added to some MPN series. To Sed and Poly mediums, Desulfomicrobium norvegicum was added as background bacterium (~106 bacteria/mL medium). This bacterium has a restricted substrate spectrum. It can function as an hydrogen scavenger for the syntrophic degradation of substrates, thereby stimulating the degradation and the growth of the prokaryotes involved. It is also quite robust, consumes oxygen rapidly (which protects other oxygen-sensitive prokaryotes), and tolerates marine salt concentrations and temperatures.
Radiotracer MPN series (Vester and Ingvorsen, 1998) were not designed to determine activity rates but aim to detect active cells with similar high sensitivity as can be obtained by fluorescence microscopy or molecular biological techniques. By the radiotracer MPN technique, growth of the prokaryotes may not be required or may be marginal, yet the technique allows the detection of metabolically active cells. MPN series were inoculated in the deep-well microtiter plates (Fig. F10) with medium to which radiolabeled 35S-sulfate (100 Bq/well) had been added. Radiolabeled sulfide formed during sulfate reduction, including FeS, S°, and FeS2, will be detected at high sensitivity by later cold distillation of hydrogen sulfide (as modified from Fossing and Jørgensen, 1989).
In order to study the reaction of the prokaryotic communities in undisturbed sediment samples to low concentrations of incoming substrates, gradient cultures were inoculated on whole pieces of sediment (Fig. F11). A mixture of monomers (4 mM each; 0.5 mL) was placed on the bottom of a glass tube and fixed with 0.5 mL agar (4% in MM at 60°C). When the agar was solidified, another 2.5 mL of agar plus 4 mL MM was added as spacer. Finally, 2 mL of a sediment subcore in 5 mL MM was placed on top, then the tube was gassed with nitrogen, closed by a rubber stopper, and cooled on ice, before incubating at the proper temperature.
Thermophiles are generally prokaryotes that grow optimally at >55°C (Topt) and have an upper temperature limit for growth (Tmax) >60°C. Most of these prokaryotes do not grow at <40°C. Thus, incubation temperatures of 50° and 60°C were used to cover different populations of thermophiles. Extreme thermophiles have a Topt > 60°C and a Tmax 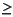 70°C, whereas hyperthermophiles have a Topt > 80°C and usually cannot grow at 60°C. Many cannot grow even at <75°C (Wiegel, 1992). Thus, an incubation temperature of 80°C was used for the hyperthermophiles.
70°C, whereas hyperthermophiles have a Topt > 80°C and usually cannot grow at 60°C. Many cannot grow even at <75°C (Wiegel, 1992). Thus, an incubation temperature of 80°C was used for the hyperthermophiles.
Several kinds of media for enrichment and cultivation of heterotrophic, chemolithoautotrophic, and chemolithorganotrophic prokaryotes were prepared on board. Cultivation media were incubated at three different temperatures: 25° (mesophiles), 50° (thermophiles), and 80°C (hyperthermophiles). Although the drilling sites do not include hydrothermal sediments, recent molecular ecological research has demonstrated that genes of anaerobic thermophiles or hyperthermophiles and extreme halophiles can be detected in shallow sediment layers (Isaksen and Jørgensen, 1994; Inagaki et al., 2001). These extremophilic microorganisms are considered to be present as dormant or relict communities. The ecological and biogeochemical significance of successfully enriched and cultured isolates will be evaluated in the context of their physiology and metabolic pathways.
Media incubated at 25°, 50°, and 80°C targeted the following physiological types of prokaryotes (numbers correspond to medium numbers in Table T5):
These enrichment media for shipboard cultivations were prepared on board and were based on MJ artificial seawater salts solution (Table T6) to which substrates were added according to the requirements of the physiological type of prokaryotes targeted. All media and incubations were anaerobic, unless otherwise stated. A 0.5- to 1.0-mL sample of master slurry was anoxically inoculated into 5 mL of media and incubated. The growth of cells was observed by microscopy. Successful enrichments were kept at 4°C until further transfers to the shore-based laboratory (JAMSTEC).
For the following enumerations and enrichments of anaerobic thermophiles, all incubations were done at a temperature of 60°C and a pH of ~8.0 and 9.0 (for exact pH at 60°C [pH60°C], see media recipes in Table T7). Some enrichments were conducted at a more alkaline pH because of a special interest in the biodiversity of alkalithermophiles. All shipboard incubations were done using the basic marine sea salt media (MSSM) (see Table T8), supplemented with the corresponding carbon sources, electron donors, and electron acceptors. Incubations that showed no growth after several weeks were heat treated (1 min at 100°C) to activate possible spores (some do not germinate during incubations at 60°C) and then were again incubated for 3 weeks for a final postcruise evaluation. The following groups were enriched at 60°C:
Acetate-oxidizing iron(III)- and manganese(IV)-reducing bacteria were enumerated by the MPN technique with acetate as the sole organic carbon source and either FeOOH or MnO2 as an electron acceptor as described by Thamdrup et al. (2000). The anaerobic, sulfate-free, bicarbonate-buffered marine mineral medium with vitamins and trace elements according to Widdel and Bak (1992) was supplemented with 5-mM acetate as electron donor and 50-mM concentration of either FeOOH or MnO2 as electron acceptor. FeOOH or MnO2 were synthesized according to Lovley and Phillips (1986, 1988). The MPN tubes for enrichment of manganese(IV)- and iron(III)-reducing bacteria were prepared before the cruise (Fe[III] red and Mn[IV] red in Table T9) and were inoculated anaerobically in tenfold dilution steps directly after sampling on board. Samples were taken from sites and depths where interstitial water gradients of manganese or iron indicated activity of manganese(IV)- or iron(III)-reducing bacteria. After the cruise, the MPN tubes are incubated at 10°C, close to in situ temperatures, for at least 1 yr. Isolation of strains and their characterization will be done from tubes with positive growth. Positive tubes will be recognized from the change in color of the precipitates from reddish brown to black (iron) and from dark brown to white (manganese) or by measuring the amount of iron(II) or manganese(II) in the medium.
Aerobic chemolithoautotrophic ammonium- or nitrite-oxidizing bacteria as well as aerobic methylotrophic prokaryotes utilizing chloride compounds have not been studied previously in deep sediments. Because of lateral seawater flow through basement rock, oxygenated seawater could penetrate into overlying deep subsurface sediment. As a consequence, ammonium could be oxidized via nitrite to nitrate by aerobic nitrifying bacteria, and methane could be consumed by aerobic methylotrophic prokaryotes. These processes are suggested by ammonium and methane profiles (see "Biogeochemistry" in the "Site 1225" chapter and "Biogeochemistry" in the "Site 1226" chapter), which show lower ammonium and methane concentrations in the bottom sediment than at medium depths. MPN counts for nitrifying bacteria were performed in liquid medium, whereas methylotrophic prokaryotes were enriched on agar plates. After inoculation, the MPNs and agar plates are incubated at 10°C for several months. For details, see Table T4.
Igneous rock samples were washed in 250-mL Pyrex bottles containing 50 mL of nitrogen-flushed marine salts solution (23.5 g NaCl and 10.8 g MgCl2·6H2O per liter). This washing step removed most of the adhering sediment particles and surface contamination. After the first washing step, the rock sample was transferred into a second wash bottle containing 50 mL of fresh nitrogen-flushed marine salts solution and was washed again by shaking and rinsing. After this step, the rock sample was collected with sterile forceps and carefully cleaned by scraping off small remaining pockets of sediment from the surface using fresh syringe needles. The loosened particles were then rinsed off with previously nitrogen-flushed marine salts solution from a syringe. Afterward, the rock was crushed into several pieces to be used for different investigations. Crushing was done either by wrapping the rock in sterile aluminum foil and breaking it into pieces with several forceful hammer strokes or by crushing the rock inside a rock crusher. This crusher is an autoclavable metal block with a well that can hold a small rock piece. A solid piston is placed into the well on the rock sample and crushes the rock with several hammer strokes aimed at the flat top of the piston.
Several pieces of the rock samples were placed into 3-M HCl for 1 min to sterilize the rock surface, rinsed with sterile marine solution, and subsequently crushed as described. These pieces were only used to inoculate media.
The pieces were stored in sterile plastic bags for thin sectioning and petrographic analysis, put into phosphate buffered saline (PBS)/50% ethanol for ion microprobe analysis, fixed for AODC), frozen at -80°C for DNA analysis, fixed in PBS/4% formaldehyde for 4´,6-diaminidino-2-phenylindole (DAPI) staining, FISH, and thin sectioning, and used for inoculation of different media. The compositions of the media are described in "Enrichments near In Situ Temperatures" in "Methods for Enrichment and MPN." In addition, to enrich for hydrogen-oxidizing endolithic prokaryotes, an autotrophic basalt medium (A-bas) was prepared (Table T4).
The A-bas was dispensed in 5-mL amounts into Hungate tubes. Half of the tubes already contained 50 µL of 1-mol/L Fe(OH)3 and 50 µL of 1-mol/L MnO2; the other half did not contain metal oxides (Thamdrup et al., 2000). The tubes were sterilized for 20 min in an autoclave at 121°C and were gassed with nitrogen while cooling on ice. Afterward, 0.15 mL NaHCO3 (1-M sterile stock) and 0.5 mL Fe(NH)4(SO4)2·6H2O (5-mM sterile stock) were added to each tube. The pH of the medium was 7.4. The tubes were inoculated with a number of basalt chips from weathered basalt (Table T10). Finally, the tubes were flushed with CO2 and 10 mL of hydrogen was added with a syringe. The tubes were incubated close to the in situ temperature for several months.
There is increasing evidence that dolomite precipitation is a microbially mediated process in which sulfate-reducing bacteria take part (Warthmann et al., 2000). Earlier studies have shown that some phospholipid fatty acids are indicative for sulfate-reducing bacteria (Parkes, 1987). Dolomite samples for phospholipid biomarker analysis were taken under clean conditions during the cold room microbiological sampling. A segment of 5-10 cm length was cut directly from the whole-round core. This segment should contain enough soft material around the hard nodule to be appropriate for a biomarker analysis. After subsampling the segment for FISH, the segment was wrapped in aluminum foil and frozen in a plastic sample bag at -20°C within 30 min. All samples were shipped frozen to the shore-based laboratory (in Zurich). Phospholipids will be extracted with organic solvents from the dolomite nodule and from the soft sediment around it. In this procedure, a solid-phase extraction is performed with dichloromethane, acetone, and methanol before saponification by mild alkaline methanolysis. The three fractions will be analyzed by GC/FID.
About 20 dolomite samples from different cores and depths were subsampled for FISH analysis. Samples were taken from the soft sediment around the hard dolomite nodules with a sterile 5-cm3 syringe, fixed in formaldehyde/PBS solution, washed, and stored in ethanol at -20°C.
Several kinds of molecular ecological analyses will be performed as postcruise investigations. The universally used approach to obtain the DNA sequences of genes of interest is polymerase chain reaction (PCR), an enzymatic reaction that produces exponentially increasing copy numbers of a target gene sequence. These multiple copies (several hundred thousand) are the prerequisite for further analysis. Molecular techniques using PCR allow rapid, sensitive, and extensive surveys of present (or past) subsurface prokaryotic communities (Marchesi et al., 1998, 2001). A plethora of molecular approaches are based, in one way or another, on PCR. Here, we list techniques that will form the basis for molecular postcruise work for Leg 201:
High-quality, nondegraded DNA and RNA of the native prokaryotic communities in deep subsurface sediments are required as starting material for PCR and for all the molecular approaches listed here (Rochelle et al., 1992, 1994). Instant deep-freezing of the sample material and storage at -80°C is essential to preserve the DNA and RNA of the native microbial community. Thawing, especially repeated freeze-thaw cycles, increases the risk of hydrolysis of high molecular weight DNA and RNA and must be avoided. The equipment and time demands of molecular community analysis, at least with the current state of technology, require that this work be done postcruise. Shipboard work was therefore limited to collecting and preserving 5-cm3 WRC samples for all parties that are interested in molecular community analysis. Recovery of living cells from deep-frozen material may be possible on a nonquantitative basis from either -80°C or liquid nitrogen; however, -20°C is not cold enough for this purpose.
As outlined in the introduction, 16S rRNA-based FISH is an important direct method to identify and count individual prokaryotic cells, based on their phylogenetic affiliation, in mixed prokaryotic communities, and environmental samples (Amann et al., 1995). In principle, a FISH profile of a prokaryotic community results in a phylogenetic and physiological profile of this prokaryotic community, as the correlation between the 16S rRNA phylogenetic framework and the physiological characteristics of particular bacterial and archaeal branches of the 16S rRNA tree is well characterized. General FISH probes for the two domains of prokaryotes, bacteria and archaea, are complemented by probes for specific bacterial and archaeal groups (e.g., probes for delta-proteobacterial sulfate-reducing bacteria, for the dominant marine cluster of sulfate-reducing bacteria, for dominant marine heterotrophs including the Planctomycetales and Cytophagales, and for methane-oxidizing archaea of the ANME-1 and ANME-2 lineages).
The protocol for FISH (Pernthaler et al., 2001) begins with a fixation step of sediment for shipboard or postcruise FISH analysis (Table T11). The cell walls of the microorganisms have to be permeabilized to the extent that oligonucleotide probes can pass through the cell walls, and at the same time the cellular ribosomal RNA content has to be preserved. This is done by fixing cells in buffered formaldehyde solution, followed by washing steps, and, at last, addition of ethanol. At this stage, the procedure can be stopped and the cells can be kept at -20°C until there is sufficient time for hybridization and microscopic examination.
The protocol for the continuation of the FISH procedure is outlined in Table T12. The first five steps describe how the samples are collected on a membrane filter and how they can be manipulated easily for hybridization and microscopy. One must adjust the sample concentration and volume empirically in order to prevent overload of the filter with sediment. An even monolayer of sediment particles and cells on the filter is desirable.
Samples were taken for shore-based geobiological analysis that will use SIMS. The purpose of this method is to analyze the stable isotopic composition of individual microbial cells (13C, 15N, and 34S), which gives important information on their biochemistry and assimilation pathways for different elements. The ion beam of the spectroscopic analysis focuses on cells or cell clusters that are immobilized on a glass slide or filter and that are identified with nonspecific fluorescence stains (DAPI/AODC) or with a phylogenetic stain (FISH). For example, combined FISH-SIMS has traced the 13C signature of methane assimilation and oxidation in methane-oxidizing prokaryotic consortia (Orphan et al., 2001). FISH-SIMS requires an ion microprobe facility, all of which are currently shore based. Therefore, shipboard work was limited to harvesting sediment samples for FISH and preserving material using different fixation and preservation protocols (Table T13).
In order to identify prokaryotic community members that are substrate limited but nevertheless active, sediment samples were preincubated with 13C-labeled substrates (glucose, acetate, and methane) before preservation and shore-based FISH-SIMS analysis. The medium chosen was a general saline solution, except for the addition of Wolfe's trace metals for methanogens. 13C-labeled tracer experiments were executed at 20 different depths (see the protocol in Table T13).
In general, the procedures for rate measurements with radiotracers involved preincubation at in situ temperature in the radioisotope van until equilibrium was reestablished. Only then was radiotracer injected into the samples and the experiments continued at in situ temperature in the isotope van for up to a month or more (if necessary, cooled samples still incubating were packed according to international regulations and shipped back to the appropriate institute by air freight). At the end of the incubation period, samples were fixed in a manner that kills the prokaryotes and preserves the radiolabeled substrate and product. Fixation ensures sample integrity and minimizes the risk of radioactive contamination during shipping and processing. At the end of the leg, these fixed samples were packed and shipped according to international regulations to postcruise investigators for further processing.
The measurement of sulfate reduction during Leg 201 is described in Table T14. The purpose was to experimentally determine the rates of bacterial sulfate reduction in subsamples of sediment taken from whole-round cores using the radiotracer 35SO42-. The use of 35S enables measurements over days to weeks with minimal disturbance of the redox equilibrium and bacterial milieu. Because of the use of radiotracer, the reduction of less than one-millionth of the sulfate can be detected, as explained in "Sensitivity of Process Rate Determinations" in "Measuring Microbial Rates and Activities" in "Introduction and Background." This typically corresponds to <1 pmol of sulfate per cubic centimeter per day (equal to a <1 nM sulfate per day), depending on incubation time and interstitial water sulfate concentration.
Measurements of methanogenesis from bicarbonate or acetate were carried out by the use of 14C-labeled substrates, applying procedures similar to those for sulfate reduction (cf. Wellsbury et al., 2000). The procedure used during Leg 201 is outlined in Table T15.
Whole-round cores from selected intervals were cleanly and anoxically sectioned, sealed in nitrogen-purged gas-tight bags with an Anaerocult A (Merck) oxygen scrubber, and stored at 4°C for shipment to postcruise investigators. The samples will be used for experimental studies of anaerobic oxidation in these sediments using 14CH4 as the substrate. The experiments will be carried out similar to those for methanogenesis (see Table T15) and incubated at in situ temperature. The conversion of the tracer substrate into carbon dioxide or biomass will be monitored.
Incubations using tritiated (methyl-3H)thymidine were initiated to measure prokaryotic DNA formation and, hence, prokaryotic growth rates. Procedures were identical to the above 14C radioisotope (Table T15) experiments except that 25 µL (650 kBq) of undiluted (methyl-3H)thymidine (3.1 MBq/mmol; Amersham, UK) was injected and incubation times were much shorter (0.5-48 hr). These experiments will be complete postcruise.
Tritiated leucine was used to measure protein production rates of prokaryotes in the sediments. Cellular protein is well correlated with total cell carbon and can therefore be used to calculate biomass production rates. Subsamples will be examined postcruise using microautoradiography to determine the fraction of the total population that incorporated the leucine. These data will be used to infer the percentage of the total cells observed that are metabolically active. The procedure used for these experiments during Leg 201 is outlined in Table T16.
Hydrogen is a key intermediate in anaerobic sedimentary prokaryotic communities. It is a product of fermentation and is utilized by sulfate reducers, methanogens, and other prokaryotes. As hydrogen is believed to be turned over rapidly, sedimentary interstitial water gradients cannot be used to infer reaction rates. Two types of incubation experiments were conducted to determine the rate of microbial hydrogen utilization. One set of experiments utilized 3H2 (see Table T17), and the other set utilized the introduction of elevated hydrogen concentrations to incubated sediments. The experiments that utilize 3H2 will be completed postcruise.
In the experiments that utilized elevated hydrogen concentrations, hydrogen was added to the headspace of a 20-mL vial that contained ~3 cm3 of a bulk sediment plug sampled with a syringe with the Luer end removed. The headspace of the vial was purged with high-purity nitrogen before the introduction of the hydrogen. The hydrogen concentration in the headspace at the start of the incubation was either 15 or 30 ppm. Samples were incubated at near in situ temperatures, and hydrogen concentration was monitored daily. Based on the change with time of the measured concentration (taking into account the approach to a new equilibrium concentration), hydrogen utilization rates were calculated. Sediment samples that were autoclaved in vials purged with nitrogen were used as controls.
Only personnel trained in radioisotope work were allowed in the radioisotope van, and all personnel in the van were required to wear disposable shoe covers, a lab coat, and safety glasses at all times. Disposable gloves were worn when handling open radioactive materials. After each experiment session, handling trays and laboratory equipment (e.g., syringes and spatulas) were monitored with a handheld surface contamination monitor (Berthold). If any contamination was present, the plastic-backed absorbent shelf paper was replaced and trays and equipment were decontaminated. A radioisotope stock log was maintained to monitor and record isotope usage. The total amounts of radioisotopes applied for radiotracer experiments during Leg 201 are presented in Table T18. At the end of the leg, samples were shipped by airfreight to appropriate institutes for further processing. All samples were packed in break-proof containers, double bagged, and packed in a sturdy International Air Transport Association (IATA)-approved container (packed and shipped as "UN2910-Radioactive material, excepted package-limited quantity of material"). Thorough (30 point) wipe tests were performed after 1 week, 3 weeks, and at the end of the expedition.
In preparation for Leg 201, a radioisotope van, manufactured according to University-National Oceanic Laboratory System (UNOLS)-approved design, was purchased and outfitted by ODP-TAMU. The van was installed on top of the Core Tech shop, on the starboard side of the JOIDES Resolution just aft of the driller's shack. The facility is a 20-ft-long steel van, equipped with two entry/exit doors (end and side) and a plumbed exterior sink. The rear one-third of the van was equipped with a cooling system that can maintain 10°C internal temperature for low-temperature sample handling. The low-temperature room includes a vent hood, counter-top working area, cabinet and drawer storage, and a nitrogen tank for sample preparation. The larger part of the van is air conditioned and houses three upright, floor-to-ceiling, adjustable temperature incubators, as well as an under-counter refrigerator and freezer. A sink (with a small isolated drained fluid capture tank) and additional cabinet and drawer storage are also installed. The van also contains a liquid scintillation counter for sample, standard, and contamination monitoring. After installation, the facility was plumbed and attached to the shipboard electrical system, phone and alarm system, and computer network. The facility is monitored with multistation contamination wipe tests on a regular basis, as well as daily personal wipe tests and routine sampling of the captured water tank. The facility remains locked at all times except when occupied by approved personnel, and access is limited to only those scientists certified by their home institution in handling radioisotopes and to members of the ODP technical staff approved by the Texas A&M Office of Radiation Safety. Protocols for spill control and decontamination procedures are outlined in Table T19.
Two depths at each of the low-activity sites (1225 and 1231), high-activity sites (1228, 1229, and 1230), and intermediate-activity site (1227) were targeted for 18O-label incubation studies. Incubations were carried out at near in situ temperatures of 4°-7°C for Sites 1225 and 1231 and at 20°-25°C for Peru margin sites. Each incubation slurry was sampled immediately after inoculation to determine starting values for water and dissolved phosphate (18OP), dissolved phosphate concentrations, and prokaryotic cell counts. These values will be compared with those from later extractions.
To avoid potential contamination by the use of heavily 18O-labeled compounds (50-99 atom% 18O) typical of biochemical approaches, incubation media were prepared using very low level 18O-labeled water (10-99 18O), sulfate, and unlabeled phosphate in various combinations. A 0.1- to 1-mL subsample of the master slurry was used as inoculum in anaerobic incubations with 10 mL of 18O-labeled media.
Three types of shipboard incubation conditions were used with phosphate:
) and unlabeled phosphate (as KH2PO4; 100 µmol/10 mL);Relatively high phosphate concentrations were required to detect the low level of 18O label in the water at the high water:phosphate ratio (105:102) used here. Additional shore-based 18O-label incubation studies will be carried out using preserved slurry and sediment samples (collected cleanly and stored anoxically at 4°C) with more heavily labeled P18O43- so that the label transfer into the water can be detected. Also, more heavily labeled H218O to permit smaller concentrations of phosphate (20-100 µM) will also be employed.
![]()