BIOGEOCHEMISTRY
The interstitial water (IW) program at Site 1226 was designed to clearly define biogeochemical zones and to allow for modeling of biogeochemical reaction rates and associated chemical fluxes. Like Site 1225, this site is characterized by active fluid flow in the underlying basement (Baker et al., 1991). The sampling strategy was similar to that applied at Site 1225, with the main difference being a higher sampling frequency of two samples per core carried through the first deep hole, Hole 1226B (total of 87 samples). After initial analyses of these samples, an additional 41 samples were collected from Hole 1226E to target specific depth intervals where interstitial water profiles displayed significant chemical variation—samples from the upper 25 mbsf (16 samples from 252.85 to 323.85 mbsf and 9 samples from 382.80 to 418 mbsf) (Table T3). In addition to routinely employed chemical analyses, we acquired IW concentration data for hydrogen, acetate, formate, nitrate, dissolved oxygen, DIC, high-resolution manganese and iron, and methane at trace levels.
As at Site 1225, microbially mediated reactions and chemical exchange across the sediment/seawater and sediment/basement interfaces drive most of the variations in interstitial water chemistry. As a result, distinct biogeochemical zones are present downhole at these sites. Profiles of dissolved species at Site 1226 are consistent with somewhat greater microbial activity compared to Site 1225.
Alkalinity and DIC have similar downhole profiles. Alkalinity increases from ~2.8 mM near the sediment/water interface to a high of ~6.9 mM at 59 mbsf (Fig. F4A). The values then decrease gradually to ~6.5 mM at 192 mbsf and more rapidly to ~2.0 mM at the bottom of Hole 1226B. The range in alkalinity at this site is ~5 mM. DIC increases from 2.8 mM near the sediment/water interface to a broad maximum of ~7.0 mM between 83 and 188 mbsf (Fig. F4B). DIC concentrations then decrease to ~2.0 mM at the bottom of Hole 1226B. The range in DIC concentrations is also ~5 mM. Both alkalinity and DIC have their greatest variability between adjacent samples in the deepest 100 m of the sediment column. The onset of this variability coincides with significant changes in lithology and physical properties associated with the transition to lithostratigraphic Subunit 1D (see "Lithostratigraphy" and "Physical Properties"). The alkalinity profile at Site 1226 is similar to the profile previously obtained at Site 846 (Shipboard Scientific Party, 1992). DIC measurements were not made at Site 846. The ~5-mM increases in alkalinity and DIC concentrations at Site 1226 are greater than the increases in these parameters at Site 1225 (see Fig. F4). This strongly suggests that rates of respiration at Site 1226 are higher than those at Site 1225.
Dissolved oxygen was not detected with the microelectrode at sediment depths of 0.1 mbsf and below. Attempts to measure oxygen in highly consolidated sediments immediately above basement failed.
Interstitial water nitrate concentrations were determined in the upper and lower sections of Holes 1226B and 1226E for comparison with Site 1225. Based on preliminary results, the pronounced zones of high nitrate at the top and bottom of the sediment column as observed at Site 1225 are not present at Site 1226. In the lowermost 16 m of Hole 1226E, nitrate concentrations reach only 3 µM, compared to 23 µM at Site 1225.
Nitrite formation is an intermediate step in nitrification, and interstitial water nitrite concentrations were analyzed for samples between 260 and 418 mbsf in Hole 1226B and between 0 and 11.95 mbsf and 401.90 and 418 mbsf in Hole 1226E and in the deepest 18 m of the site (Table T3; Fig. F4). In an attempt to detect active turnover of interstitial water nitrogen species, interstitial water nitrite concentrations were determined within 5 to 30 min after sampling. Nitrite concentrations range from 0.0 to 0.23 µM, with the highest values between 290 and 350 mbsf (Fig. F4). Nitrite concentrations roughly track nitrate concentrations in the lowermost ~20 m.
The high-resolution dissolved manganese profile (Fig. F4D) shows considerable structure, including features of potential significance to subsurface microbial activity. Unlike at Site 1225, manganese concentrations >30 µM are present just below the seafloor (0.1-0.2 mbsf). After a slight drop and a second peak at 1.8 mbsf, dissolved manganese declines to 5 µM at 80 mbsf. Concentrations drop less precipitously over the next 80 m to 2 µM. At 250 mbsf, dissolved manganese rapidly increases to form a peak, with values exceeding 36 µM at 303 mbsf (Sample 201-1226B-33X-2, 135-150 cm). Concentrations then decrease to a local minimum of 18 µM at 360 mbsf and increase to a high of 44 µM near the base of the hole.
The upper part of the dissolved manganese profile is somewhat similar to that at other open-ocean sites that underlie regions of primary productivity (Brady and Gieskes, 1976). The shallow concentration peak probably results from the dissolution of manganese oxide phases driven by microbial manganese reduction. The changes in gradients at 80 and 160 mbsf are likely caused by manganese precipitation. The deeper part of the profile probably necessitates a complex explanation because the dissolved manganese peaks likely signify reduction of solid manganese oxides—phases that would not accumulate in the presence of significant amounts of sedimentary organic carbon. Dissolved organic carbon may be diffusing downward and reacting with solid manganese phases at depth. This dissolved manganese profile at Site 1226 may represent a special case where an interval of low organic sedimentation (and presumably high burial rate of authigenic manganese oxides) was followed by an interval of relatively high primary productivity in surface waters (and presumably rapid accumulation of organic carbon).
Similar to Site 1225, dissolved iron concentrations at Site 1226 (Fig. F4E) show considerably more scatter between adjacent samples than do other analyzed species. Concentrations are not significantly different between true replicate samples, indicating that the scatter is not caused by poor analytical precision. At Site 1225, two explanations were offered for this variability: (1) oxidation of iron in some samples during squeezing or water handling or (2) inclusion of solid iron in some samples. To address the first possibility, most interstitial water samples at Hole 1226B were squeezed through a three-way valve into a second glass syringe evacuated of air. In general, however, adjacent samples from Hole 1226B show more variation in iron concentration than samples processed by conventional means from Hole 1226E, possibly because of iron adsorption on glass. To address the second possibility, several samples were pushed through 0.45-µm (standard) and 0.10-µm filters. However, iron concentrations for all of these dual analyses lie within analytical precision (Table T4). If solid iron contributes to the variation in "dissolved iron," the corresponding particulates must be <0.10 µm in size. Other than the unresolved scatter, the iron profile is characterized by three short intervals of high concentrations. These peaks are centered at ~10, 315, and 385 mbsf. These peaks are present on the shoulders of peaks in dissolved manganese.
Dissolved sulfate concentrations decrease from near seawater concentrations at the sediment surface to 19.8 mM at 246.20 mbsf (Table T3; Fig. F4F). Below this depth, sulfate increases to 25.2 mM at 417.20 mbsf, just above the basaltic basement. The downhole profile generally exhibits lower sulfate values than that at Site 1225. Moderate sulfate reduction occurs throughout most of the sediment column at Site 1226.
The downhole profile of dissolved strontium is similar to that obtained for Site 846, although with much higher depth resolution. Concentrations are similar to that of seawater (86 µM) in the uppermost sample at 0.10 mbsf and then steadily rise to values >310 µM at 205 mbsf (Fig. F4G). Below 300 mbsf, concentrations sharply decrease to 190 µM above basement. The overall profile suggests an active zone of carbonate recrystallization in lithostratigraphic Subunit 1C (see "Description of Lithostratigraphic Units" in "Lithostratigraphy"), with diffusive exchange with seawater at the top and with modified seawater at the bottom.
Dissolved lithium concentrations track those determined for Site 846, although there is an offset at low values that requires additional attention (Fig. F4H). Concentrations are similar to that of seawater (26 µM) in the uppermost sample at 0.37 mbsf and then steadily decrease to a low of 11 µM at 220 mbsf. With a slight concavity at ~300 mbsf, concentrations increase to ~23 µM above basement. Lithium is removed from interstitial water during carbonate recrystallization. As with strontium, the overall profile suggests an active zone of carbonate recrystallization in lithostratigraphic Subunit 1C.
Concentrations of dissolved barium are below the detection limit (0.1 µM) in all samples analyzed from Site 1226.
Concentrations of dissolved calcium, potassium, and magnesium were determined by inductively coupled plasma-atomic emission spectroscopy. Interstitial water samples were diluted at 50:1 with deionized water, and standard curves were constructed by diluting International Association for the Physical Sciences of the Ocean standard seawater aliquots with a range of deionized water volumes. Given current operating conditions for the instrument, the dilution is probably not ideal and this may have affected analytical precision, especially for magnesium and potassium.
The downhole profiles of dissolved calcium, potassium, and magnesium are similar to those obtained for Site 846, although with far greater vertical resolution. Concentrations of calcium are similar to that of seawater (10.8 mM) in the uppermost sample at 0.10 mbsf and then decrease to 7.5 mM at ~100 mbsf (Fig. F4I). Dissolved calcium steadily returns to 10.8 mM between 360 and 380 mbsf, an interval where silica concentrations drop precipitously and the sediment contains chert horizons. Calcium concentrations then rise steeply to 16 mM over the lowermost 40 m. Dissolved potassium generally decreases from 11.8 mM at the seafloor to 10.6 mM at 330 mbsf. Below this depth and after a possible offset to higher concentrations, potassium drops rapidly to 9 mM above basement. By contrast, dissolved Mg concentrations steadily decrease from 54 mM near the seafloor to 48 mM above basement.
Total dissolved sulfide (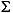 H2S = H2S + HS-) is present at detectable levels (>0.0002 mM) at depths greater than 0.59 mbsf (Fig. F4J). The concentrations of sulfide increase linearly with increasing depth to 0.43 mM at 21.45 mbsf. Below this depth, the concentrations of sulfide continue to increase with increasing depth, but the data show more scatter. The scatter in the sulfide data corresponds to the increase in scatter in the methane data at the same depths (Fig. F4K). Like methane, sulfide may have been lost from samples through degassing. In this case, the maximum values best approximate in situ concentrations. Sulfide concentrations continue to increase with increasing depth and form a broad maximum of 0.5-0.7 mM between 45 and 178 mbsf. Peak concentrations of sulfide reach 0.7 mM at 74 mbsf. Below 178 mbsf, the concentrations of sulfide decrease linearly with depth to 0.089 mM at 235 mbsf. Between 235 and 279 mbsf, sulfide concentrations vary between 0.09 and 0.19 mM before decreasing to values of <0.0001 mM at depths below 281 mbsf.
H2S = H2S + HS-) is present at detectable levels (>0.0002 mM) at depths greater than 0.59 mbsf (Fig. F4J). The concentrations of sulfide increase linearly with increasing depth to 0.43 mM at 21.45 mbsf. Below this depth, the concentrations of sulfide continue to increase with increasing depth, but the data show more scatter. The scatter in the sulfide data corresponds to the increase in scatter in the methane data at the same depths (Fig. F4K). Like methane, sulfide may have been lost from samples through degassing. In this case, the maximum values best approximate in situ concentrations. Sulfide concentrations continue to increase with increasing depth and form a broad maximum of 0.5-0.7 mM between 45 and 178 mbsf. Peak concentrations of sulfide reach 0.7 mM at 74 mbsf. Below 178 mbsf, the concentrations of sulfide decrease linearly with depth to 0.089 mM at 235 mbsf. Between 235 and 279 mbsf, sulfide concentrations vary between 0.09 and 0.19 mM before decreasing to values of <0.0001 mM at depths below 281 mbsf.
Collectively, the dissolved sulfide and sulfate profiles (Fig. F4F) suggest a broad zone centered between 25 and 150 mbsf where sulfate is reduced to sulfide. The presence of sulfide between 235 and 279 mbsf is consistent with the small inflection in the sulfate profile at 250 mbsf and further indicates a deeply buried zone of sulfate reduction. The broad maximum in sulfide corresponds to the maxima in the metabolic products DIC (Fig. F4B) and ammonium (Fig. F4L). In contrast to DIC and ammonium, the low solubility of metal sulfides results in the complete removal of sulfide at 281 mbsf. This depth approximately coincides with a transition toward higher dissolved iron concentrations downhole (Fig. F4D).
Ammonium concentrations rise from 22.4 µM at the top of the section to a maximum of 641 µM between 90 and 121.2 mbsf then decline more gradually downward, reaching 222 µM at 410 mbsf (Fig. F4L). The ammonium profile is similar to that obtained previously at Hole 846C (Shipboard Scientific Party, 1992). The shape of the ammonium profile is similar to the alkalinity and DIC profiles and mirrors the sulfate profile (Fig. F4F). Increased interstitial water ammonium, alkalinity, and DIC and decreased sulfate are consistent with the degradation of organic matter via bacterial sulfate reduction.
Concentrations of dissolved phosphate generally decrease from 8 to ~2 µM between 1.3 and 125 mbsf (Fig. F4P). Below 125 mbsf, phosphate declines smoothly, reaching a value of ~1 µM at 360 mbsf. Between 382.8 and 390.8 mbsf, phosphate concentrations are only ~0.3 µM.
Concentrations of acetate and formate were analyzed in 40 interstitial water samples from Hole 1226B (Table T3; Fig. F4M, F4N). Concentrations were generally higher than those at Site 1225 (see "Biogeochemistry" in the "Site 1225" chapter) and exceeded the detection limit in the majority of samples. Three distinct depth intervals can be distinguished: (1) concentrations of both compounds are below or around the detection limit between the sediment surface and 116.2 mbsf; (2) concentrations of both acetate and formate range between 0.4 and 1.2 µM between 116.2 and 274.7 mbsf; and (3) maximum concentrations of acetate and formate, from 1.1 to 4.3 µM and 0.7 to 2.2 µM, respectively, are present below 298.1 mbsf. The acetate profile has two distinct peaks at 336.50 and 409.6 mbsf, with values of 3.9 and 4.3 µM, respectively.
The stepwise increase in acetate and formate concentrations below 116.2 mbsf coincides with a sharp decline in total organic carbon (TOC) from ~0.5% to 0.1% (Shipboard Scientific Party, 1992) and the transition to lithostratigraphic Subunit 1B (see "Description of Lithostratigraphic Units" in "Lithostratigraphy"). The depth interval from 116.2 to 274.7 mbsf also coincides with the plateau of maximum methane concentrations (Fig. F4K; see discussion below) and minimum sulfate concentrations. Presently, the significance of increasing acetate levels with increasing burial depth remains unclear. Wellsbury et al. (1997) showed that interstitial water acetate concentrations generally increase with temperature in sediments from the Blake Ridge. Temperature at the bottom of Site 1226 sediment reaches ~25°C (see "In Situ Temperature Measurements" in "Downhole Tools").
Methane was detected in all samples from Hole 1226B (Table T5; Fig. F4K). No other hydrocarbons were detected. Maximum methane concentrations exceed 2 µM in the sediment horizon from ~80 to 300 mbsf. These concentrations are an order of magnitude higher than the maximum concentration found at Site 1225 in the same depth horizon. Notably and unlike at Site 1225, methane concentrations do not drop to zero above the sediment/basement interface.
Consistent with observations at Site 1225, the two procedures employed for extracting the dissolved methane led to different recoveries. On average, methane concentrations were slightly higher in samples that were extracted for 24 hr at room temperature than in samples extracted with the standard ODP technique used for safety purposes (Fig. F4K).
Incubations for hydrogen analysis were conducted on 13 samples from Hole 1226B and 4 samples from Hole 1226E (Table T6). The samples were incubated at temperatures close to the in situ sediment temperatures (4°, 13°, and 21°C) (see Table T6 for details). The temperatures were chosen based on the temperature profile modeled from the temperature tool data (see "Downhole Tools").
Hydrogen concentrations in the incubations from Site 1226 are low (<1 nM) (Fig. F4O) compared to those at Site 1225 and measured by Hoehler et al. (1998) in shallow nearshore sediments. Concentrations ranged between 0.13 and 0.74 nM without any clear downhole trends. A more detailed consideration of the controls on hydrogen concentrations requires an analysis of free energies of the dissimilation reactions that involve hydrogen.
The dissolved interstitial silica profile at Site 1226 generally increases with depth over the upper 300 mbsf (Fig. F4Q), probably due to dissolution of amorphous biogenic silica (opal-A). The highest measured concentrations (1300-1400 µM) are present in lithostratigraphic Subunit IB between 275 and 315 mbsf. Overall, the downhole silica profile tracks variations in color reflectivity (see "Color Reflectance
Spectrophotometry" in "Lithostratigraphy"), with maxima corresponding to diatom-rich intervals. The pronounced decrease between 360 and 400 mbsf coincides with the presence of chert nodules and layers consisting of quartz (see "Mineralogy" in "Lithostratigraphy"). Below this interval of low silica, concentrations sharply increase, although the onset of this rise cannot be resolved because of poor recovery. Toward basement, the concentrations drop as expected by the flow of silica-poor basement water.
Chloride concentrations increase from 553 mM near the sediment/water interface to 561 mM at a depth of 40.2 mbsf (Fig. F4R). This trend is most likely due to the diffusion of chloride from high-chlorinity glacial seawater out of the sediment column (e.g., McDuff, 1985). The values are relatively constant to a depth of 390.8 mbsf and then decrease to 552 mM at 413 mbsf. This near-basement decrease has at least two possible origins. Lower-chlorinity basement water is consistent with basement waters being derived from interglacial bottom waters. However, the diagenesis of chert in this part of the sediment column could also release water.
Contents of TOC, calcium carbonate, total nitrogen, and total sulfur were not determined throughout most of the sediment column at Site 1226. However, they were determined on selected samples from representative intervals of Subunits IB (50-120 mbsf) and ID (260-310 mbsf) (Table T7). Samples were chosen to compare their chemical composition in relation to lithology, which alternates between nannofossil ooze and diatom ooze throughout Subunit IB and the upper part of Subunit ID (see "Unit I" in "Description of Lithostratigraphic Units" in "Lithostratigraphy"). Total nitrogen and total sulfur concentrations are generally low or below detection limit. Notable differences in TOC content are present in adjacent layers of nannofossil ooze and diatom ooze, with the latter type commonly containing higher amounts of TOC and lower amounts of carbonate. Both the carbonate and TOC concentration data of Site 1226 are in agreement with data from respective intervals obtained at Site 846 (Shipboard Scientific Party, 1992) and provide a potential explanation for high-amplitude variation of TOC concentrations in the Site 846 data set. TOC and calcium contents throughout the sediment column were measured at Sites 851 and 846. In the upper 150 mbsf at Site 846, TOC typically exceeds 0.5%, with several values above 1% and a few approaching 2% (Shipboard Scientific Party, 1992). The average TOC contents of Site 846 are higher than those at Site 851. Maintenance of higher microbial activity by these higher TOC contents may account for alkalinity and DIC being higher at Sites 846 and 1226 than at Sites 851 and 1225.
