SITE 1227
Site Description and Lithology
Site 1227 is located on the Peru margin at 8°59.46´S, 79°57.34´W, ~100 m from the location of ODP Leg 112 Site 684 in the Trujillo Basin, which is a small, fault-bounded pond of sediments. Water depth at this location is 427.5 m. Presently, this site is positioned within the upwelling system. The sediment succession consists of four lithologically distinct units and contains a mixture of marine and terrestrial components (Suess, von Huene, et al., 1988; D'Hondt, Jørgensen, Miller, et al., 2003).
The age of Unit I sediments (0–11.95 mbsf) is <0.9 Ma. They are mostly laminated and consist of diatom-bearing silt and clay-rich diatom ooze with occasional foraminiferal ooze layers. Unit I was deposited under a strong upwelling regime. Unit II (11.1–34.1 mbsf), of Pleistocene age, contains dark olive, bioturbated, silty sediments with glauconite and phosphate layers that indicate possible winnowing of sediments during the times of sea level lowstand (Suess, von Huene, et al., 1988). Variations in the amount of terrigenous input for Unit II and evidence for sediment winnowing by bottom currents are attributed to fluctuations in sea level throughout the depositional history of Unit II. Unit III (34.1–53.1 mbsf), of Pliocene age (Suess, von Huene, et al., 1988), is characterized by reduced diatom abundance and a high percentage of clay, feldspar, and quartz, which implies the presence of higher terrigenous input in this unit. Vertically graded sequences are common. Overall, the unit exhibits a fining-upward trend; sediments are bioturbated. A strong porosity minimum, accompanied by grain density and magnetic susceptibility maxima, is observed in the interval between 40 and 50 mbsf. Unit IV (53.1–151 mbsf), of late Miocene age, is separated from the overlying Unit III by a large unconformity; its duration spans 5.7–8.7 Ma (Suess, von Huene, et al., 1988). The sediments of Unit IV are mostly dark green clays and nannofossil-bearing ooze. Volcanic ash layers and volcanic shards occur through the unit. Laminated dolomite layers are present. Overall, this unit is characterized by a lower terrigenous input than the three stratigraphic units above it. It was also least affected by bioturbation and reworking by currents. Estimated sediment accumulation rates are 20 m/m.y. for the Quaternary sequence, 30 m/m.y. for the Pliocene, and 50 m/m.y. for the Miocene (Suess, von Huene, et al., 1988).
Biogeochemistry of Pore Water
The concentrations of major metabolites and electron acceptors in the pore fluids of Site 1227 indicate the presence of active microbial communities in these sediments (D'Hondt, Jørgensen, Miller, et al., 2003). Sulfate concentrations decrease downcore from seawater values to below the detection limit at ~40 mbsf (Fig. F4). Methane concentrations increase steeply below 38 mbsf.
DIC concentrations reach a maximum of ~25 mM at 38 mbsf, in the sulfate–methane transition zone. Below this horizon, DIC decreases to 20 mM and concentrations remain constant through the rest of the sediment column recovered. Concentrations of pore water ammonium increase steadily with depth (Fig. F5), reaching 23 mM at the bottom of the profile. At the boundary between Units II and III (36–38 mbsf), the gradient of ammonium concentrations shows a pronounced kink in data from Hole 1227A, with a steep gradient in Unit III below a smaller gradient in Unit II. Results from Hole 1227D do not indicate a distinct kink but have lower sampling resolution. Core recovery in the interval between 43 and 72 mbsf was poor (D'Hondt, Jørgensen, Miller, et al., 2003), which might have led to contamination of some samples with seawater. However, sulfate concentrations (Fig. F4) show little evidence of such contamination, clearly <5%. The ammonium profile obtained during Leg 112 (Suess, von Huene, et al., 1988) also exhibits a change in the gradient at the same depth horizon. In contrast to ammonium, chloride concentrations increase steadily with depth, with no significant kinks in the profile (Fig. F6). The location of the kink coincides with the interval of sulfate–methane transition. Sediments of Unit III, just below the kink, have lower porosity than in adjacent horizons.
Elemental and Nitrogen Isotope Composition of Sedimentary Organic Matter and Pore Water Ammonium
Sediments from Site 1227 are rich in organic matter, which is typical for highly productive upwelling regions (Fig. F7; Table T3). Concentrations of organic carbon are 5–9 wt% and total nitrogen 0.4–0.6 wt%. Adsorbed ammonium constitutes 1%–10% of TN (calculated as discussed for Site 1230). Atomic C/N ratios vary between 12 and 21. Relatively high values of sedimentary C/N ratios are consistent with episodic input of terrestrial organic matter, suggested by the sedimentological evidence. The overall trend of C/N increasing with depth through the upper 35m may indicate possible preferential remineralization of N-containing compounds.
Sedimentary 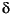 15N values for these lithologic units vary (Fig. F7; Table T3). Nitrogen isotope ratios in Unit I (last 0.9 m.y.) change from 6.8
15N values for these lithologic units vary (Fig. F7; Table T3). Nitrogen isotope ratios in Unit I (last 0.9 m.y.) change from 6.8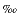 to 4 downcore. Sediments of Units II and III (Pliocene) have 15N values between 2 and 4. Unit IV (Miocene sediments) is characterized by significantly heavier isotopic values, at ~9–10.
to 4 downcore. Sediments of Units II and III (Pliocene) have 15N values between 2 and 4. Unit IV (Miocene sediments) is characterized by significantly heavier isotopic values, at ~9–10.
Nitrogen isotope ratios of pore water ammonium show a different pattern (Fig. F7; Table T3) and far less variation than solid phases. Ammonium 15N is 3–4 heavier than sedimentary 15N in the upper 36 mbsf (Units I and II) and becomes progressively lighter from ~8 near the sediment/water interface to ~5 at 36 mbsf. The isotopic composition of ammonium in pore water of Units III and IV is lighter than sedimentary 15N and decreases from 5 in Unit III and the upper part of Unit IV to ~4.5 downcore. In Unit III, the ammonium isotopic composition is fairly close to the sedimentary 15N; in Unit IV, ammonium is ~5, which is 4 lighter than the sedimentary 15N (Fig. F7).
Sources and Sinks of Pore Water Ammonium
Pore water biogeochemistry and the structure of ammonium concentration and isotopic composition at Site 1227 are very complex. Increasing concentrations of pore water ammonium downcore usually indicate continuous degradation of organic matter at depth. However, at Site 1227, the pore water chemistry is influenced by the presence of a hypersaline subsurface brine of Miocene age below 40–50 mbsf (Suess, von Huene, et al., 1988), as manifested by increasing concentrations of Mg2+, Ca2+, Na+, and Cl– with depth (Suess, von Huene, et al., 1988; D'Hondt, Jørgensen, Miller, et al., 2003).
A nearly linear relationship between NH4+ and Cl– concentrations (Fig. F8) below 36.95 mbsf suggests that the predominant source of ammonium diffusing toward this horizon is the brine, rather than decomposing organic matter. Above the 36.95-mbsf depth horizon, ammonium is probably added by decomposition of organic matter, as indicated by concave-down shape of the plot for the upper interval. The kink in the ammonia profile near 37 mbsf shows up on Figure F8 as a local minimum. Undoubtedly, the kink is partially attributable to the relatively low porosity layer near the top of Unit III, which should require a steeper gradient for ammonium to balance fluxes. However, the gradient for chloride should also steepen, and Figure F8 should indicate linearity through this region if both solutes behave conservatively and the system is in steady state. As noted above, sulfate shows little indication that the samples were significantly diluted with seawater during recovery, and the good agreement of shipboard and USC ammonium measurements is evidence that the kink is not a result of analytical problems. If the system is in steady state, Figure F8 is strong evidence that the kink reflects a local sink for ammonium, separating deep sources (predominantly the brine) and shallower sources from decomposing organic matter.
The localization of sink near this horizon might be reasonable, as it lies in the sulfate–methane transition horizon, where pH changes occur and new mineral phases, such as carbonates, are likely to precipitate (Meister et al., this volume). Struvite (MgNH4PO4) could be a candidate phase (Elderfield et al., 1981). Laboratory experiments demonstrated coprecipitation of struvite with Mg-bearing carbonate minerals (Pontoizeau et al., 1996; Rivadeneyra et al., 2006). But degree of struvite saturation at the ambient pH of ~7 in these pore waters is <0.001. Consequently, it is unclear what phase might provide a sink in this horizon.
Nitrogen isotopes provide some additional insights into the nature of sources and sinks that must exist. A useful approach is to construct a mixing diagram, where 15N of ammonium is plotted against inverse concentrations, [NH4+]–1 (Fig. F9), as described by Faure and Mensing (2005). On such diagram, conservative mixing between two localized ammonium sources with different isotopic compositions and concentrations would result in a straight line connecting the 15N values of the end-members, if the profile is in a steady state. Deviations from a linear relationship indicate additional sources or sinks through the region sampled. The shape of the curvature depends on the isotopic composition of the nitrogen added (or lost) and the depth region in which it is added. If diffusion is the dominant transport mechanism and steady state exists, a tangent to the data points, extrapolated to the y-intercept, indicates the composition of the net upward ammonium flux.
From the mixing diagram (Fig. F9), we can identify four features that correlate with the sources and sink identified based on the concentration profiles and the ammonium vs. chloride. Three sources are associated with different lithologic units: (1) ammonium in Unit IV diffusing upward from the brine with 15N of ~4.4 as indicated by projected intercept, (2) ammonium with isotopic composition of ~8 added through the Units I and II, and (3) a small and not readily apparent ammonium source in Unit III that adds 15N of ~6. Finally, the sink in the kink region alters the relationship so that the extrapolated tangent indicates the upward flux has become isotopically lighter. This requires that the sink preferentially removes 15N. In addition, ammonium is lost at the sediment/water interface.
In Unit IV, the nearly linear trend formed by the data points indicates predominantly conservative mixing between ammonium diffusing from the brine and ammonium released in Unit III (Fig. F9). The data from Units I and II, on the other hand, strongly deviate from the simple mixing relationship illustrated by the dotted line in Figure F9. The steady-state interpretation requires continuous production of heavier ammonium along the mixing path. The region inside the gray circle is strongly influenced by the sink near 37 mbsf, which affects isotopic composition as well as concentration.
The isotopic composition of the required sources and the sink can be estimated by calculating mass balances. A two-layer reaction-diffusion model (for 14NH4+ and 15NH4+) was constructed with the assumption of a steady-state condition (detailed description of formulations used in this model is given in Prokopenko et al. [2006]) and applied to calculate diffusive ammonium fluxes toward and away from the 36.95-mbsf horizon. Isotopic composition of the fluxes was calculated as follows:
-
15Nflux = [(15J/14J)/Rstd – 1) x 1000,
where J is the flux of 15NH4+ and 14NH4+, respectively, Rstd is 15N/14N ratio in the atmospheric N2 gas. Advective transport was ignored, as the Peclet number is >>1 (Table T2). Fluxes were calculated as first derivatives of the concentrations according to Fick's first law. Flux leaving Unit III, just below the 36.95-mbsf horizon, is 0.56 ± 0.20 µmol/(cm2yr) with isotopic composition of 5.8, while the upward flux from this horizon is 0.14 ± 0.05 µmol/(cm2yr) with 15N of 0.9 (see Fig. F10). Therefore, our calculations show that ~75% of ammonium may be lost in the vicinity of the 36.95-mbsf horizon, if the ammonium profile is currently in the steady state. The loss is accompanied by small positive isotopic fractionation with the fractionation factor of approximately +3.0 ± 0.5. This small positive isotopic fractionation associated with the apparent sink would point toward a nonbiological mechanism, as biological ammonium uptake is usually accompanied by large-magnitude negative isotopic fractionation (Hoch et al., 1992).
The interpretation of the ammonium sources, as well as the sinks suggested by the changes of slopes on the mixing diagram, strongly depends on whether the represented system in the steady-state condition. We limit the following discussion to the upper 40 mbsf (Units I and II), since below this horizon the major source of ammonium is the Miocene brine, and factors controlling its 15N are presently unknown.
Nitrogen Budget at Steady State in Units I and II
Assuming a steady-state condition and applying the reaction-diffusion model (Prokopenko, 2004), we calculated the magnitude of the ammonium fluxes at several depth horizons through the upper 37 mbsf of the sediments and relative contribution of the ammonium flux from below (0.14 µmol/[cm2yr]) to the total flux at each of these horizons. Then, 15N of total ammonium at each horizon was calculated as the isotopic mass balance between the 15N of flux from below 36.95-mbsf horizon (5.5, observed) and a heavier ammonium with constant isotopic composition added through Units I and II. The best fit to the data points was obtained with 15N of the heavier end-member of 8.5 (Fig. F9, solid line).
The average isotopic composition of the bulk sedimentary nitrogen in the upper 36.95 mbsf of the sediments is 3.8. This is ~4.7 lighter than the 15N of 8.5 of ammonium flux added to the pore water through this interval. So, the question relevant to the present study is what is the mechanism of the apparent isotopic enrichment of pore water ammonium relative to the organic nitrogen in the upper 36 mbsf of the sediments at Site 1227?
If the ammonium concentration and 15N profiles are in steady state, then the enrichment of ammonium in heavier isotopes relative to Norg signifies a loss of the isotopically heavier nitrogen from organic matter through Units I and II. In this case, as diagenesis progresses, the preserved organic matter should become isotopically lighter. Indeed, measured 15N of sedimentary organic nitrogen in Units I and II (Fig. F6) becomes progressively lighter downcore, changing from 6.8 at the surface to ~4 in the lower part of Unit I, and reaching values between 2.5 and 3 within Unit II. Consequently, if the ammonium profile at Site 1227 is in steady state, diagenetic processes at this site result in the 15N shift of ~4 in preserved organic nitrogen. Isotopically heavier ammonium might be released via one of the following mechanisms: (1) preferential degradation of isotopically heavier organic N (Macko and Estep, 1984), that might be more labile; (2) preferential assimilation of isotopically lighter ammonium into bacterial biomass (Lehmann et al., 2002); or (3) preferential decomposition of an isotopically heavier, more labile marine fraction of organic matter relative to a more refractory terrestrial component (Prokopenko et al., 2006; Sweeney and Kaplan, 1980). High C/N ratios in the sediments of Site 1227 imply that significant component of terrestrial organic matter is present, although the sediment supply may come from settings that are currently relatively arid. The 15N of the isotopically heavier ammonium end-member of 8.5 is consistent with the marine origin of the source organic matter. Therefore, the latter scenario could be responsible for the observed isotopic enrichment of pore water ammonium relative to the sedimentary nitrogen, if the system is in steady state.
The steady-state reaction-diffusion model of Prokopenko (2004) was used to predict the 15N of pore water ammonium, which would result from mixing between the brine source and ammonium contributed from decomposition of organic matter occurring with the fractionation factor of 2. The 15N of ammonium released at 2.85 mbsf (our shallowest data point and a "zero" depth in the model) was set to be 8.5. The results are plotted on the mixing diagram as a dashed line and indicate that while this process does cause a deviation from the conservative mixing relationship, it cannot fully account for the observed enrichment of pore water ammonium relative to the Norg.
Finally, we calculated the ammonium production in the interval between 2.85 and 36.96 mbsf. Applying the approach described for Site 1230, we find that present-day ammonium flux produced throughout this interval cannot be supported by the sedimentation flux of Norg with the N concentration equal to the topmost measured interval concentration, which is 0.31 wt%. The ammonium production flux is ~1.1 ± 0.15 µmol/(cm2yr), while Norg accumulation flux at 2.85 mbsf is 0.22 ± 0.05 µmol/(cm2yr) (calculated from average porosity 0.8 and concentration of N = 0.31 wt%). The unsupported ammonium flux, most likely, results from the variability of the weight percent of organic nitrogen through time, or, in other words, the nonsteady-state depositional history of the sediments at Site 1227.
Evaluating our assumption of the steady-state condition at Site 1227, we conclude that the steady-state scenario leaves two problems unresolved: (1) our estimated ammonium production flux exceeds our estimate of the organic N burial flux at 2.85 mbsf, and (2) the isotopic composition of ammonium predicted by our steady-state model substantially deviates from the observed data (Fig. F9).
Nonsteady-State Scenario
An alternative scenario is that the ammonium profile and its isotopic composition represents a nonsteady-state condition, where 15N of ammonium is strongly affected by the contribution from the decomposition of recently deposited organic matter with 15N > 8. If a pulse of organic-rich material with the heavier 15N has been deposited within the last several thousand years, under the regime of intense postglacial upwelling, a corresponding pulse of isotopically enriched ammonium should be released into the pore water. This would create an isotopically distinct diffusive front that would propagate downward.
At present, the isotopically heavier ammonium from the pulse of organic matter deposited during the last 10 k.y. would have influence to a depth of ~30 mbsf if
-
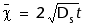 ,
,
where
-
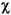 = mean distance of diffusion,
= mean distance of diffusion,
-
Ds = 8.12 x 10–6 cm2/s is the diffusion coefficient of ammonium in the sediments with porosity = 0.8, and
-
t = time.
In this case, the isotopic enrichment of pore water 15N relative to 15N of Norg would be due to the addition of the new, isotopically heavier end-member rather than fractionation during the decomposition of organic matter.
With an average sedimentation rate of 20 m/m.y. estimated for Site 1227, the upper 2 m of the sediment column would represent ~100 k.y. Our first isotopic measurement was done at a depth of 2.85 mbsf, so 15N of the modern surface sediments is unknown. In a region ~2° south of Site 1227, the reported isotopic composition of modern organic matter is between 6 and 8 (Libes and Deuser, 1988). At a site ~4° south of Site 1227, Ganeshram et al. (2000) reported sedimentary values of 15N for the last 100 k.y. that vary between 6 and 11. From those studies, we can infer the values of 15N of the organic matter deposited in this region to be most likely between 8 and 10. In this case, isotopic composition of ammonium through the upper 36.95 mbsf of the sediment column at Site 1227 may be dominated by mixing between ammonium of 8–10 released in the upper 1–2 mbsf and ammonium coming from the brine (with isotopic composition of ~5). If this recently deposited material had higher reactivity than the older material, it could have a pronounced effect on the mass balance for both ammonium and its 15N.
The nonsteady-state hypothesis would offer an alternative explanation for the change in the concentration gradient at 37 mbsf. It seems possible that the apparent coincidence of the methane/sulfate boundary and the diffusing ammonium pulse both represent the influence of the low porosity zone, which would retard downward transport of both sulfate and the nonsteady-state pulse of recently added ammonium. Further measurements of near-surface sediments are required test this hypothesis. Finally, a nonsteady-state influence on ammonium behavior would not preclude application of steady-state models to other pore water solutes because the response time would depend on the length scale of the solute in question. Sulfate, for example, has a much shorter length scale and should have reached nearly steady-state distribution during the past 10 k.y.
