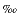 enriched in the lighter isotope, 14N, over the heavier isotope, 15N.
enriched in the lighter isotope, 14N, over the heavier isotope, 15N.
Organic matter that is deposited and buried deeply in the seabed is the main carbon source for subseafloor microbial life. It is also the primary energy source available for the deep biosphere at the sites studied during Leg 201. No other energy sources were identified that could have an importance of comparable magnitude to that of buried organic carbon. Rates of mineralization and microbial activity, as quantified by modeling of major pore water constituents, relate closely to the organic contents of the sediments (D'Hondt, Jørgensen, Miller, et al., 2003; D'Hondt et al., 2004). Total organic carbon (TOC) was measured throughout the sediment column at all sites, with high-resolution sampling near the surface at selected sites (Meister et al., this volume b). TOC contents vary as much as a hundred-fold between the equatorial Pacific sites and the Peru margin sites. At open Pacific Site 1225, TOC contents are very low, 0.0–0.2 wt%, whereas at Site 1226 they reach 1–2 wt%, within the upper 150 meters below seafloor (mbsf). In the Peru Trench (Site 1230) TOC contents are higher (mostly 2–3 wt%) but not as high as might be expected from the depletion of pore water sulfate already at 9 mbsf. The steep sulfate depletion at Site 1230 appears to be driven by methane ascending from deeper deposits rather than by high organic mineralization rates in the upper meters of the sediment.
On the Peruvian shelf, high phytoplankton productivity caused by upwelling supplies the underlying sediments with abundant organic matter. TOC values here scatter in the range of 1–12 wt%. Particularly in Pleistocene–Holocene deposits at the shallower sites, TOC data indicate systematic variations that have been interpreted as glacial–interglacial cycles and may be associated with eustatic sea level variations (Wefer et al., 1990). These variations in carbon concentrations along with variations in carbon isotopic compositions and lignin contents probably reflect variations in marine productivity and diagenetic overprinting rather than variations in terrigenous inputs (Louchouarn et al., this volume). Sediment dating by accelerator mass spectrometry (AMS) 14C measurements of bulk organic matter reveal that the Holocene section is largely absent at Peru shelf Site 1227, but sediment from the Pleistocene deglaciation is almost 6 m thick (Skilbeck and Fink, this volume). At Sites 1228 and 1229 the Holocene section is 2–2.5 m thick, whereas the upper Pleistocene section is thin and possibly eroded. A gradual increase with depth and time in the atomic C/N ratio of organic matter from 12 to >16 within the same late Pleistocene–Holocene time window at Site 1227 demonstrates the result of microbial degradation on residual organic material (Prokopenko et al., this volume).
Through degradation of buried organic matter a part of the degradation products accumulate in pore water and can be detected as a pool of dissolved organic carbon (DOC). This pool consists of a complex mixture of organic molecules with large diversity in molecular weight and chemical composition (Burdige, 2002). During further microbial mineralization, the remaining DOC becomes increasingly refractory to enzymatic attack. Smith (this volume) analyzed bulk concentrations of DOC in pore water samples from Leg 201 cores using a high-temperature catalytic oxidation method (Sharp et al., 2002). DOC concentrations mostly showed limited variations of 0.2–0.5 mM at the open Pacific sites and 0.5–12 mM at the Peru margin sites. Surprisingly, DOC concentrations were very high in the subsurface sediment column of the Peru Trench (Site 1230), reaching a flat maximum of 20 mM at 50–150 mbsf (Smith, this volume). This DOC concentration is so high that a yellow coloration was clearly visible in freshly collected pore water (Shipboard Scientific Party, 2003d [Site 1230]) and a strong ultraviolet light absorption at 325 nm could be measured.
Fehn et al. (submitted [N1]) reviewed the iodine isotopic system as a tracer of organic material buried subsurface and included new data of Snyder et al. (submitted [N2]) from Site 1230. Iodine is strongly associated with organic material; therefore enrichment of the cosmogenic radioisotope 129I with a half-life of 15.7 m.y. can be used to indicate the age of buried organic material. Pore fluids from Site 1230 are strongly enriched in iodine (Martin et al., 1993) and show a distinct decrease in the ratio of 129I to total I (127I) with depth, from 920 x 10–15 in surface sediments to 300 x 10–15 below 100 mbsf. Fehn and coworkers calculated ages of 40–60 Ma for the pore fluids, which appear surprisingly old considering that they are derived from subducting sediments in an active margin. Whereas at Site 1230 sediments of Miocene age are subducting below Holocene–Pleistocene deposits, the source sediments for iodine, and possibly for some of the methane, thus appear to be of Eocene age or even older and may be stored as a large and very old reservoir within the overriding arc (Fehn et al., submitted [N1]).
Very little is known about the compound classes that compose DOC in deep subsurface sediments. In near-surface coastal and ocean-margin sediments total dissolved carbohydrates (DCHOs) represent 10%–40% of the total DOC (see Burdige, 2002, for a review). Burdige (this volume) analyzed DCHOs in Leg 201 pore water samples from all sites and found concentrations ranging from 0 to ~1500 µM C. Interestingly, there were no consistent downhole trends but there were distinct differences in concentration levels between sites. These differences do not relate to sediment organic content, however. Pacific Sites 1225 and 1231, as well as Peru Trench Site 1230, have the highest DCHO concentrations overall, whereas the Peru shelf sites with high organic content have low DCHO concentrations. Although there is presently not a clear interpretation of these concentration differences, it is apparent that several factors controlling production and degradation of DCHOs interact to determine DCHO concentrations.
Amino acid concentrations in the pore water similarly did not show systematic trends with increasing depth at any of the sites. The concentrations showed differences in the concentration ranges between sites with the highest concentrations found on the Peru margin and in the Peru trench Site 1230 (Mitterer, this volume). Aspartic acid, glutamic acid, serine, and glycine were detected and occurred in concentrations mostly in the range of 1–10 µM. With geological age, the remaining amino acids in sediment organic material may undergo a slow racemization from the predominant L-forms to D-forms (Mitterer, 1993). An alternative source of D-forms is the degradation of bacterial cell walls in which the structural biopolymer, peptidoglycan, contains D-aspartate, D-glutamic acid, D-serine, and D-alanine (e.g., Schleifer and Kandler, 1972; Lomstein et al., in press). The D/L ratios of amino acids, however, did not show systematic increases with depth, and, thus, neither racemization nor an increasing contribution from bacterial cell walls could be demonstrated (Mitterer, this volume).
Methane concentrations in sulfate-depleted sediments strongly exceed 1 bar of partial pressure in cores from Peru margin Sites 1227, 1229, and 1230. Consequently, the supersaturated methane formed gas voids in the sediment cores upon retrieval and some of the gas was lost. In order to retain the gas and obtain realistic concentration data, a pressure core sampler (PCS) was used successfully for coring at selected depths at Site 1230, which has very high in situ methane concentrations and also methane clathrates (Dickens et al., 2003). Methane concentrations as high as 959 mmol/kg were measured at 150 mbsf, the greatest depth sampled using the PCS. Measuring methane in samples collected using the PCS requires that the gas pressure be gradually released from the core and the volume of released gas measured at 1 bar.
As PCS operations are rather time consuming, an independent approach for determining high methane concentrations was tested during Leg 201 (Spivack et al., this volume). The method is based on immediate sampling of gas from voids within the decompressed core liner and simultaneous measurement of CH4, N2, and Ar in the collected gas. As in situ concentrations of the conservative gases, N2 and Ar, can be estimated from their environmental concentrations and solubilities, the ratio of CH4:N2 or CH4:Ar can be used to backcalculate the in situ partial pressure of CH4. This method allows calculation of in situ methane concentrations in sediment obtained by normal coring operations with the advanced piston corer (APC). Preliminary testing of the method provided concentrations of several hundred millimol CH4 per kilogram of sediment, consistent with data from PCS cores.
During core retrieval, piston cores that contain high concentrations of gas and gas hydrates undergo distinct changes in physical properties that can be monitored and thereby provide information about the gas properties. A temperature, pressure, and conductivity (TPC) tool mounted at the face of the standard ODP APC was successfully tested at Site 1226 (Ussler et al., this volume). Future applications of this tool at gas hydrate sites will show how ascent curves of TPC data may be used to interpret gas geochemistry.
Organic matter in deep subsurface sediments reflects mineralization that has taken place over thousands or millions of years during burial since the organic material was originally deposited. The general changes in structure and composition with depth and age is overprinted by chemical alterations of the organic matter and by synthesis of new biomass of deep biosphere prokaryotes. Because of the extremely low energy supply and slow growth, growth yield and thus biomass production are expectedly only a small fraction of the organic matter turned over. Because the deposition rate of organic matter to the seafloor may have changed considerably over the millions of years during which burial took place, it is difficult to backcalculate the cumulative amount of organic degradation since the time of burial.
Prokopenko et al. (this volume) assumed for Peru Trench Site 1230 steady-state conditions of sedimentation, burial, and diagenesis over the last >1 m.y. in order to calculate the amount of particulate organic nitrogen (PON) that had degraded to ammonium over that time window. They modeled the modern pore water profile of ammonium and arrived at the very plausible conclusion that 35%–42% of PON had been ammonified. The produced ammonium is isotopically very similar to the PON, on average only 0.7 enriched in the lighter isotope, 14N, over the heavier isotope, 15N.
A similar backcalculation of organic mineralization for Peru shelf sediments is complicated by several factors. The influx of organic matter has varied significantly over glacial–interglacial cycles, presumably due to stronger upwelling and thus higher phytoplankton productivity in interglacial periods (Altabet et al., 1995; Ganeshram et al., 1995). Sediment organic carbon on the shelf originates from both marine and terrestrial sources, which have different carbon and nitrogen isotopic compositions; terrestrial organic nitrogen is isotopically lighter than the marine source.
Prokopenko et al. (this volume) modeled sedimentary total organic nitrogen (TON) and ammonium concentrations and their N isotopic compositions for Site 1227 under both steady-state and nonsteady-state assumptions. The goal was to understand controls on diagenesis and diffusion transport in relation to the Holocene–Pleistocene history of deposition and mineralization. The results of their very innovative modeling efforts demonstrate the complexity of the nitrogen cycle and its geological evolution but also offer mechanisms to explain observed geochemical and isotopic data. It appears likely that a pulse of marine organic matter with 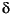 15N of 8–10 deposited during the present interglacial is currently being degraded at a relatively high rate, thus releasing ammonium that is enriched in 15N relative to underlying residual bulk organic nitrogen that has 15N of ~3. Through diffusional exchange, this isotopically heavy ammonium is currently affecting the 15N of ammonium in Pleistocene–Pliocene subsurface sediment at 10–35 mbsf, below which the signal is diluted by the pool of ammonium that diffuses upward, with 15N of 5, from the underlying Miocene brine. These results thus provide interesting information about progressing organic mineralization over geologic time.
15N of 8–10 deposited during the present interglacial is currently being degraded at a relatively high rate, thus releasing ammonium that is enriched in 15N relative to underlying residual bulk organic nitrogen that has 15N of ~3. Through diffusional exchange, this isotopically heavy ammonium is currently affecting the 15N of ammonium in Pleistocene–Pliocene subsurface sediment at 10–35 mbsf, below which the signal is diluted by the pool of ammonium that diffuses upward, with 15N of 5, from the underlying Miocene brine. These results thus provide interesting information about progressing organic mineralization over geologic time.
A broad spectrum of biogeochemical processes associated with microbial degradation of buried organic matter and transformations of gases, inorganic ions, and mineral phases was studied in sediment cores of Leg 201. Shipboard chemical analyses focused primarily on pore water species that were sampled at high depth resolution at all sites drilled. These data were published in the Leg 201 Initial Reports volume (D'Hondt, Jørgensen, Miller, et al., 2003). During the leg, only a few cations with particular relevance to the main objectives of the cruise were analyzed routinely (Fe2+, Mn2+, Ba2+, and, partly, Sr2+). Other major cations (Na+, K+, Ca2+, Mg2+, and Sr2+) were analyzed postcruise by inductively coupled plasma–atomic emission spectroscopy (ICP-AES) (Donohue et al., this volume). Whereas concentrations of these cations generally show only small variations with depth at the open Pacific sites, a strong and consistent trend of increasing concentrations with depth is observed on the Peru shelf as the result of brine at depth (Kastner et al., 1990; D'Hondt, Jørgensen, Miller, et al., 2003).
Pore water profiles of the main substrates and products of microbial processes provide information on progressing mineralization of organic material and biological energy metabolism. D'Hondt et al. (2004) used a biogeochemical flux model to calculate net reduction rates of nitrate, oxidized metals, and sulfate based on pore water concentrations of nitrate, manganese, iron, sulfate, and sulfide. At the ocean-margin sites and the most active open-ocean site, sulfate reduction is overall the dominant mineralization pathway. At the lowest-activity open-ocean Site 1231, sulfate reduction is marginal and organic carbon mineralization is mainly driven by reduction of manganese and iron. Nitrate reduction is detectable only at low-activity Sites 1225 and 1231. Interestingly, at Sites 1225 and 1231 nitrate penetrated into the sediment column not only from seawater above but also from fluids seeping through basaltic crust below.
Rates of biogeochemical processes were also measured experimentally in retrieved core samples by incubation with radiolabeled substrates. Tracers included 35S-labeled sulfate and 14C-labeled bicarbonate or acetate, and the results provide information on gross rates of sulfate reduction and methanogenesis in deep subsurface sediments. As an example, results of such sulfate reduction measurements from Site 1226 are presented in Figure F2A. Reduction rates drop steeply over the uppermost 10 m from 20–30 pmol/cm3/d at the sediment surface to <1 pmol/cm3/d. Below 20 mbsf rates are mostly below the detection limit of 0.3 pmol/cm3/d. This limit is set by the minimum of radiolabeled sulfide that must be formed during incubation experiments to be distinguishable from background in the liquid scintillation counter. Detectability of sulfate reduction was optimized by long incubations of several weeks and by improved separation of radioactive sulfide and sulfate after incubation (Kallmeyer et al., 2004). The sulfate reduction data above background in subsurface sediments are very scattered; there is distinct clustering of detectable rates in the depth interval 280–310 mbsf. The sulfate profile through the entire sediment column at Site 1226 shows a subsurface decrease from 29 mM to a minimum of ~21 mM at mid-depth (Fig. F2B). Below that, sulfate concentrations again increase toward seawater value near basement. Methane is detectable throughout the sulfate zone, although at very low concentrations of 0–3 µM (Fig. F2B). The enhanced sulfate reduction rates measured at 280–310 mbsf coincide with a distinct maximum in dissolved manganese and thus in manganese reduction (Fig. F2C). These increased sulfate reduction rates also coincide with a minimum in color reflectance, possibly due to light-absorbing manganese oxides (D'Hondt, Jørgensen, Milller, et al., 2003). Sediment at this depth was deposited during the Miocene "carbonate crash" ~10 m.y. ago, at a time of low organic deposition and thus more efficient burial of manganese. This coincidence of enhanced mineralization rates shows that modern subsurface activity is related to past oceanographic conditions at the time of sediment deposition. The reasons for enhanced sulfate reduction in the manganese-rich zone are, however, not clear. H2S concentrations show a broad peak at sediment depths (20–280 mbsf) where there is little sulfate reduction (Fig. F2C). Manganese distribution clearly exerts an equally important role on free sulfide distribution, as does the rate of H2S production.
Based on the pore water sulfate profile at Site 1226, D'Hondt et al. (2004) modeled the sulfate flux, from which a mean sulfate reduction rate of 0.02 pmol/cm3/d can be calculated for the upper 200 mbsf (see below). This modeled rate is an order of magnitude below the minimum detection limit for experimental sulfate reduction measurements and explains why below 10 mbsf most data fall below detection (Fig. F2A). It should be noted that the model approach provides net sulfate reduction, whereas experimental rate measurements provide gross sulfate reduction. Generally, differences between the two may be caused by (a) net rates underestimating sulfate reduction due to reoxidation of sulfide to sulfate in the subsurface sediment or (b) gross rates overestimating sulfate reduction because of a stimulation of microbial activity by coring and sample handling.
Pore water sulfate profiles clearly show the very different levels of microbial activity in the subsurface sediments of the open-ocean and ocean-margin sites. As the temperatures of all sediments drilled (1°–26°C) are within the normal range of psychrophilic or mesophilic microorganisms, no thermochemical sulfate reduction takes place. Microbial sulfate reduction is associated with a kinetic isotope effect in which sulfate with the lighter isotope, 32S, is reduced faster than sulfate with the heavy isotope, 34S (Kaplan and Rittenberg, 1964; Chambers and Trudinger, 1979). Seawater sulfate has had a nearly constant isotopic composition of 34S = 21 (relative to the international Vienna Canyon Diablo Troilite [V-CDT] standard over the past 50 m.y. (Paytan et al., 1998). Biological sulfur fractionation during sulfate reduction is 15–40 in pure cultures of sulfate-reducing bacteria, whereas in modern marine sediments the isotopic difference between sulfate and sulfide is generally somewhat larger, 30–60 (Canfield, 2001a, 2001b) but may reach as high as >70 (Wortmann et al., 2001; Rudnicki et al., 2001). Some of the earliest evidence that sulfate reduction takes place through viable microorganisms in deep subsurface sediments was in fact derived from stable sulfur isotope studies that showed biological isotope fractionation (Zak et al., 1980).
The sulfur isotopic compositions of sulfate from Leg 201 sites clearly show the effect of biological isotope fractionation associated with microbial sulfate reduction (Böttcher et al., this volume). The deviation in isotopic composition relative to 34S in seawater varies in proportion to the degree of sulfate depletion: no deviation is detectable at Peru Basin Site 1231, where sulfate concentrations remain nearly constant through the entire sediment column, open Pacific Sites 1225 and 1226 show modest deviation, and strongest deviation, 34S of as much as 80û90, occurs at the Peru margin sites where sulfate is completely consumed at depth. Using a mathematical model of Claypool (2004) that takes both sulfate reduction and diffusive exchange into account,
Böttcher et al. (this volume) calculated sulfur isotope fractionations by microbial sulfate reduction of 14–40 (i.e., in the lower range for modern marine sediments).
Seawater sulfate has isotopic diversity not only in sulfur but also in oxygen. During sulfate reduction in laboratory cultures or in marine sediments residual sulfate becomes enriched in the heavy isotope 18O (Mitzutani and Rafter, 1973; Fritz et al., 1989; Böttcher et al., 1998; Aharon and Fu, 2000). In contrast to the kinetic isotope effect for sulfur fractionation by microbial sulfate reduction, the primary mechanism of oxygen isotope fractionation is thought to be an intracellular isotope exchange between water and sulfur intermediates formed during dissimilatory sulfate reduction. In marine sediments this leads to partial oxygen isotope equilibration between sulfate and water molecules in the pore fluid. Complete oxygen isotope equilibration would imply enrichment of 18O in pore water sulfate from 18O of 9.5 in seawater sulfate to >30 (relative to Vienna standard mean ocean water [V-SMOV]) (Böttcher et al., 1998). An alternative pathway of 18O enrichment in pore water sulfate could be a reoxidation of sulfide by Mn(IV) or Fe(III), possibly via disproportionation reactions, whereby the produced sulfate incorporates pore water oxygen (Böttcher et al., 2001).
Blake et al. (this volume) analyzed oxygen isotope compositions of pore water sulfates from the open Pacific sites of Leg 201. The results demonstrate a clear relationship between degree of microbial sulfate reduction and oxygen isotopic enrichment in residual pore water sulfate. 18O-SO42– does deviate from seawater value at Site 1231, where sulfate depletion is minimal, whereas pore water sulfate is 10–20 enriched in 18O at Sites 1225 and 1226. Surprisingly, calculated 18O isotope enrichment for Sites 1225 and 1226 ranges from 42 to 79, which is several-fold higher than the highest fractionations yet recorded in laboratory cultures of sulfate-reducing bacteria. The cause of this strong fractionation is not known but may be an unexpectedly high degree of intracellular equilibration with oxygen from water during extremely low cellular sulfate reduction rates. Additionally, it could be a result of sulfide oxidation and sulfur disproportionation, whereby the produced sulfate incorporates oxygen from pore water (Böttcher et al., 1998, 2001). The maximum 18O-SO42– enrichments observed remain below the expected value for complete isotope exchange equilibrium with ambient pore water.
As an example of these isotopic fractionations and their relation to sediment diagenesis, Figure F3 shows data on pore water chemistry and 34S and 18O of sulfate from relatively organic rich eastern equatorial Pacific Site 1226. Dissolved inorganic carbon (DIC) and ammonium are produced by mineralization of sediment organic matter. Maximum concentrations are reached below 50–100 mbsf. Toward the crust that underlies the 418-m sediment column, concentrations decrease again as DIC and ammonium diffuse down into crustal fluid of near-seawater composition. The ammonium maximum in the upper part of the sediment column occurs where turnover of organic nitrogen is higher than deeper in the sediment. This peak coincides with a peak in H2S produced from sulfate reduction. Sulfate concentrations decrease from the seawater value of 29 mM to a minimum of 21 mM at mid-depth (200–300 mbsf), whereas 34S-SO42– increases to a maximum of 37. Toward the basaltic crust sulfate concentrations increase again to a near-seawater value of 26 mM while 34S-SO42– similarly decreases to 25. This trend toward seawater values at the crust shows that fluid flow through basalt has 34S-SO42– similar to seawater, which indicates that crustal sulfate has not been significantly affected by bacterial reduction along its flow path. These interpretations of sulfur isotope distributions are confirmed by 18O-SO42–, which reaches +28 at 70–140 mbsf and drops again to near-seawater values at the bottom of the sediment column (Fig. F3). On the Peru shelf, in contrast, sulfate diffusing upward from the underlying Miocene brine is enriched in 34S relative to seawater. This deep sulfate source has apparently already been modified by microbial sulfate reduction, probably at the time of brine formation by evaporation.
![]()