MICROBIOLOGY
Over the last two decades, studies of the deep subseafloor biosphere have mostly focused on the magnitude of microbial populations as measured by total microscopic cell counts (Parkes et al., 1994, 2000). Such data have revealed that the subsurface biosphere is the largest reservoir of biomass on Earth (Whitman et al., 1998). Yet, only few studies have addressed the structure, diversity, and function of subsurface microbial communities. It was therefore one of the primary objectives of Leg 201 to explore physiological and phylogenetic types of bacteria, both with respect to overall diversity and to dominant populations. Discrimination between active and dormant organisms was a particular challenge because only metabolically active cells currently contribute to biogeochemical processes.
During Leg 201 and during subsequent extensive laboratory studies a range of approaches was used to quantify bacterial abundance, diversity, and activity. These approaches have included
- Total microscopic cell counts;
- Cultivation methods, mostly starting with the most probable number (MPN) technique;
- DNA- and RNA-based techniques, in particular the establishment of sequence information;
- Key genes and the use of fluorescence in situ hybridization (FISH); and
- Radiotracer and stable isotope tracer experiments on specific microbial processes.
Because successful application of all these approaches depends fully on recovery of deep sediment samples uncontaminated by microorganisms from the surface world, tests for contamination were conducted during the entire coring operation and during sampling for microbiology.
Core and Sample Handling and Contamination Tests
The sedimentary environment in the deep subseafloor has experienced highly constant conditions for millions of years. Prokaryotic organisms that inhabit this stable ecosystem are therefore expected to be sensitive to chemical and physical changes induced by core sampling and handling, in particular to changes in oxygen, temperature, and pressure. While drilling in the tropics during Leg 201, warming of cores during retrieval through warm surface waters and during handling on deck was of major concern. Another concern was release of hydrostatic pressure, >50 MPa, in cores retrieved from the maximal water depth (5086 m) of Site 1230 in the Peru Trench. The technology for subsampling cores without decompression was, however, not yet available at the time of Leg 201.
Deep biosphere research requires that recovered cores are suitable for microbiological study and that recovered core material is not contaminated with microbes from drilling fluid. Standard ODP coring and sampling procedures were therefore modified in order to minimize changes after coring (Shipboard Scientific Party, 2003a). Intermittent warming was reduced by immediately transferring core sections to a cold room, where all subsequent microbiological sampling was done. Exposure to oxygen and to contaminating bacteria was reduced by maintaining core segments as whole-round cores until sampling. Core segments were cut under aseptic and anoxic conditions, and only sediment from the uncontaminated center of cores was used for enumeration, isolation, and identification of microorganisms or for activity analyses and experiments. The new core handling procedures applied during Leg 201 caused a delay in coring and a loss of stratigraphic continuity in archived cores, yet it was an absolute prerequisite for obtaining high-quality material for microbiological studies. However, each site that was drilled during this leg had been drilled and described before according to standard ODP procedures (during DSDP Leg 34, Peru margin ODP Leg 112, and equatorial Pacific ODP Leg 138) (Fig. F1).
An indication of whether potential contamination with microorganisms from the surface world has taken place may be obtained by comparing the diversity of sediment populations with those in the seawater applied for drilling. Such a comparison, based on 16S ribosomal RNA (rRNA) gene sequencing and culturing methods, have been used to argue against contamination of samples taken from deep granitic aquifers (Pedersen et al., 1997). During Leg 201, more direct contamination tests were made in all samples used for microbiological studies and for activity measurements in order to check for potential introduction of liquid or cells from the outside. Two types of contamination tracers were applied during drilling: (a) a water-soluble perfluorocarbon tracer (PFT) and (b) fluorescent microspheres (Smith et al., 2000a, 2000b; House et al., 2003). The fluorocarbon (perfluoromethylcyclohexane, C6F11CF3) was continuously fed into the drilling fluid at tracer concentration and could subsequently be detected at high sensitivity in core samples potentially affected by this diffusible contaminant. The fluorescent microspheres were of similar size (0.5 µm) as indigenous microorganisms (0.2–1.3 µm) and tested potential penetration of microbial cells from the outside into the core samples. About 1011 microspheres were injected at the tip of the APC as it released so that the core surface became smeared with this particulate tracer. The principal results of these two contamination tests were that the applied APC coring and handling procedures can produce sediment samples with nondetectable particle contamination and with dissolved tracer contamination corresponding to <0.1 µL seawater or <50 prokaryotes/g sediment. The latter is a maximum estimate, as it assumes that bacterial cells can follow diffusion of PFT tracer into the cores. The results also show that extended core barrel (XCB) coring generally produces contaminated samples and that continuous and scrupulous contamination tests are a prerequisite for obtaining microbiological samples with minimal risk of contamination. As phylogenetic identification of subseafloor communities develops in the future it may become possible to detect contaminating cell types from the surface and apply this as an additional test of potential contamination.
The most widely used procedure to count prokaryotic cells in marine sediments is based on staining with a fluorescent nucleic acid stain followed by microscopic counts of fluorescent cells. Such counts using the stain acridine orange have been made on a wide range of ODP sediment cores, including cores from the Peru margin and the equatorial Pacific Ocean (Parkes et al., 1994). Data from these authors demonstrate an exponential decrease of microbial cell densities with depth, from ~108 cells/cm3 at the sediment surface to two orders of magnitude lower counts below 100 mbsf. Minimum cell densities of 104–105 cells/cm3 were consistently detected even in the deepest sediments sampled at 800 mbsf.
The acridine orange direct count (AODC) method was applied at all sites of Leg 201 and yielded cell densities that are consistent with the general trend of the global data set of Parkes et al. (2000). Yet, cell numbers also demonstrate deviations that could be interpreted in terms of changing past oceanographic conditions at the time of sedimentation or in terms of the present geochemical zonation of the sediment column. Figure F4 shows cell enumeration data from all Leg 201 sites compared to previously censused sites. The figure includes data from 1 mbsf to the deepest sediment cored. Cell densities from open-ocean sites generally fall below the geometric mean of the global data set, whereas cell densities from the Peru margin fall on or above the mean. This trend is in accordance with previous observations that show greater bacterial populations in ocean-margin sites than in open-ocean sites (Parkes et al., 2000) and also large populations associated with gas hydrate sites (Wellsbury et al., 2000; Cragg et al., 1996).
These results lead to several important conclusions. The community size in different oceanic settings reflects the burial rate of organic carbon, which is the main energy source for the prokaryotic cells. Thus, cell densities are highest where concentrations of metabolic products (DIC, CH4, and NH4+) and net rates of sulfate and iron reduction are highest. Some of the highest cell densities ever observed beneath the seafloor were detected in sediments recovered from the Peru shelf. In contrast, the open-ocean sites contain some of the lowest average cell densities ever observed in deep-sea sediments (D'Hondt et al., 2004). The community size decreases by several orders of magnitude between the surface sediment and the deep subsurface, yet cell density rarely drops below the detection limit of 104–105 cells/cm3. Even in the deepest sediment from 420 mbsf (Site 1226) or in the oldest sediment of ~35 Ma (Site 1231), morphologically intact cells with stainable DNA could be counted.
It is striking that cell numbers from the continental shelf sites fall closer to the geometric mean of the global data set than do cell numbers from the open-ocean sites. This indicates that the data set may be skewed toward the ocean margin relative to the global distribution of sediments. In fact, ODP coring sites are far more abundant along the ocean margins than in the central gyres of the open ocean. This means that global extrapolation of total subsurface bacterial biomass is probably overestimated and should be corrected to take into account the areal coverage of sediments with lower cell densities. Due to the sparsity of data for open-ocean and low-productivity provinces, such a geographically weighted extrapolation remains uncertain and calls for further cell enumerations in poorly represented ocean regions.
Cultivation of Prokaryotes
It is somewhat discouraging to realize that most bacteria that live in natural environments have so far resisted all efforts to bring them into laboratory culture. This may, at least in part, be explained by differences in environmental conditions offered during cultivation in defined media relative to the complex conditions of the natural habitat. In the habitat, energy substrates are generally present in very low concentrations, but they are steadily produced. In cultivation media, the energy substrate is present in high concentration from the start in order to support a significant number of cell divisions. The cultivation media thereby select for opportunistic organisms able to grow relatively fast under rich substrate conditions that they would never encounter in the deep subsurface.
A most striking quality of the deep biosphere is the extremely slow growth of organisms, with estimated mean generation times of many years (Whitman et al., 1998; Schippers et al., 2005). It has not yet been possible to measure such low growth rates by direct approaches, so current estimates are based on a comparison of total population size with potential metabolic rates, calculated from chemical data or from sensitive radiotracer experiments (D'Hondt et al., 2002). The exceedingly slow growth of subseafloor populations may help explain why the number of successful cultivations and isolations has been quite limited (e.g., Bale et al., 1997; Barnes et al., 1998; Inagaki et al., 2003; Mikucki et al., 2003). It also raises the question to what extent physiology and growth characteristics of isolates that have indeed been obtained from deep sediment cores are representative of those deep subseafloor populations. Although this question is difficult to answer at the present time, isolation and characterization of indigenous bacteria from ODP core material does provide useful information for at least part of the microbial populations.
A large number of cultivation experiments were initiated during Leg 201 in order to study taxonomic and physiological diversity of subsurface prokaryotic communities. These cultivation experiments used many different selective media for enrichments in order to target a broad spectrum of physiological types with respect to energy metabolism and adaptation to the physical-chemical environment. Some cultivations focused on heterotrophs that could use different electron acceptors for respiration or that could degrade different monomeric or polymeric carbon sources. Other cultivations focused on methanogens and acetogens or on fermenting or spore-forming prokaryotes. Still others targeted chemoautotrophic prokaryotes utilizing different combinations of inorganic redox couples as electron donors and acceptors. Incubation conditions were chosen to cover a broad spectrum of adaptations to pH, salinity, or temperature (psychrophiles, mesophiles, and thermophiles, respectively).
Sediment samples were homogenized into slurries using cultivation media and subsequently diluted in tenfold steps in media that could potentially support growth of specific physiological types of prokaryotes. By scoring the highest dilution steps with positive growth, the MPN of viable cells in the original samples could be estimated according to a standard statistical procedure (American Public Health Association, 1989). Such MPN counts typically provide only a minimum estimate of true numbers of organisms that are viable in situ because many prokaryotes (or even the vast majority) are not cultivable with currently available methods. Shipboard MPN cultivations also served as starting material for later enrichments and isolations of the prokaryotes in shore-based laboratories. Isolation of prokaryotes from the highest dilutions with positive growth maximizes the chance of ultimately cultivating organisms that are quantitatively dominant and that are, therefore, biogeochemically important.
Bacteria were successfully cultured and isolated from multiple depths at every site. The highest cultivation success was obtained at Peru margin Site 1229, where MPN counts reached as high as 48,000 cells/cm3 (H. Cypionka, pers. comm., 2005). This corresponds to a counting efficiency of up to 0.1% of the total cell counts obtained by AODC (D'Hondt et al., 2004). Such counting efficiency is not significantly lower than MPN efficiencies obtained in marine surface sediments. It was important by these counting and cultivation experiments that the media had low concentrations (>100 µM) of mixed substrates. Such low-nutrient media have been found to yield higher numbers of cultured bacteria and also higher diversity (e.g., Kaeberlein et al., 2002; Zengler et al., 2002). In fact, bacteria adapted to low substrate concentration may be irreversibly damaged by exposure to high substrate concentrations (Barer and Harwood, 1999). Isolations furthermore utilized mixed substrates and potential trace compounds by adding sediment extract or reducing agents to the media.
Cultivation results indicate that living bacteria are present throughout the entire range of subseafloor depths sampled (D'Hondt et al., 2004). Thus, at open Pacific Sites 1225 and 1226, bacteria could be isolated from the sediment surface to the greatest depth cored at 307 or 420 mbsf, corresponding to sediment ages of ~10 or 15 Ma, respectively. A total of 172 pure cultures were isolated by the microbiology group at Oldenburg University in Germany (H. Cypionka and B. Engelen, pers. comm., 2005). Interestingly, a majority of the strains turned out to be facultative aerobes, even though they were isolated from anoxic environments using strict anaerobic technique. There is presently no clear explanation for this observation. The 16S rRNA genes were analyzed for all the isolates and show that these represent a broad spectrum within the bacterial kingdom. The isolates belong to the following six lineages: 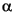 -,
-, 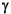 -, and
-, and 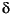 -Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes. About 40% of all the isolates could be assigned to the spore-forming genus Bacillus. Most of the isolates are related to known marine organisms. Others are distant from known organisms and will be assigned to new genera. It was an interesting observation that the 16S gene of one isolate from open-ocean Site 1225 differed from its nearest known relative within Bacteroidetes by as much as 14%. It thus appears to represent a new genus of a distinct phylogenetic lineage.
-Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes. About 40% of all the isolates could be assigned to the spore-forming genus Bacillus. Most of the isolates are related to known marine organisms. Others are distant from known organisms and will be assigned to new genera. It was an interesting observation that the 16S gene of one isolate from open-ocean Site 1225 differed from its nearest known relative within Bacteroidetes by as much as 14%. It thus appears to represent a new genus of a distinct phylogenetic lineage.
Whereas all of those isolates grow in the mesophilic temperature range,
Biddle et al. (this volume) attempted to isolate psychrophilic methanogens and heterotrophic bacteria by incubating sediment at 2° or 10°C. Samples were taken from Site 1230 in the Peru Trench at 5300 m water depth where the sediment is rich in organic matter and gas hydrates occur below the 9-m-deep sulfate zone. One of the objectives was to determine whether metabolically active methanogens are present in the gas hydrate zone. Several previous studies have searched for archaeal populations in subseafloor sediment but did not find 16S rRNA genes of methanogens in their libraries (Biddle et al., 1999; Marchesi et al., 2001; Mikucki et al., 2003; Newberry et al., 2004). More recent studies, however, have indeed found evidence for methanogens in subseafloor sediment by using specific 16S rRNA or functional gene (methyl coenzyme-M reductase; mcrA) primers (Marchesi et al., 2001; Newberry et al., 2004; Sørensen et al., 2004; Parkes et al., 2005; Inagaki et al., 2006). A mesophilic methanogen species designated Methanoculleus submarinus was recently isolated from 247 mbsf in the Nankai Trough (Mikucki et al., 2003). Although a few psychrophilic methanogens are in pure culture, none are from marine subsurface sediment (Chong et al., 2002; Franzmann et al., 1997). Interestingly,
Biddle et al. (this volume) found during initial enrichments in aerobic medium an archaeal polymerase chain reaction (PCR) product by ribosomal intergenic spacer analysis (RISA) fingerprinting that belongs to a group of uncultivated benthic Crenarchaea (Ventriani et al., 1999; Bowman and McCuaig, 2003). The basic physiology of this group remains unknown, and further attempts of isolation were not successful. The group appears commonly in the 16S rRNA gene libraries obtained from Leg 201 sites (Sørensen et al., 2004; Inagaki et al., 2006).
All the main substrates for methanogenesis were applied in the low-temperature enrichments by
Biddle et al. (this volume), yet methanogens could not be detected, even after 2 yr of incubation. Upon enrichment in marine broth medium, however, several isolates of heterotrophic bacteria were obtained that are moderately psychrophilic in their temperature growth range. These represent the genera Photobacterium, Shewanella, Halomonas, and Vibrio. These produce extracellular lytic enzymes, protease or esterase, that are active at low temperature and may lyse macromolecular organic compounds such as proteins or lipids (Boetius and Lochte, 1994; Luna et al., 2004). The Photobacterium isolate has high 16S rRNA sequence similarity to the previously described P. profundum (Nogi et al., 1998) and similarly shows adaptation to high hydrostatic pressure by growing at 40 MPa (the highest pressure tested), corresponding to 4000 m water depth (Biddle et al., this volume).
At the other end of the temperature scale, a new group of thermophilic bacteria was isolated from sediments of the Peru margin. Altogether 10 thermophilic isolates were obtained from sediment cores taken from 1–2 mbsf at in situ temperatures ranging from 2° to 12°C (Lee et al., 2005). Attempts to isolate them from greater sediment depths were not successful. The bacteria are anaerobic heterotrophs belonging to family Thermoanaerobacteriaceae and have growth optimum temperatures of 64°–68°C. A novel genus, Thermosediminibacter, was proposed for the described isolates. Given the high temperature required for their growth (>40°ñ–50°C) and their apparent absence from deeper, older sediments, the occurrence of these bacteria in shallow and relatively cold shelf sediments can possibly be explained by their dispersal through the ocean from hotter environments, sedimentation onto the seafloor, and maintenance of viability for the few thousands or tens of thousands of years represented by the uppermost 1–2 m of sediment at these sites.
Isolation of novel genera of bacteria in combination with the discovery of unknown deeply rooted archaeal 16S gene sequences shows that previously undiscovered prokaryotes exist in deep subseafloor sediments of the ocean. It remains an important task of future Integrated Ocean Drilling Program (IODP) research to develop further approaches for cultivating of these unknown microorganisms and for studying their physiological and biochemical adaptation to their special environment.
DNA- or RNA-Based Identification
Since cultivation-based analyses of subseafloor microbial populations are very time consuming in comparison to the number of successful isolations, cultivation-independent molecular analyses have become an indispensable part of all population studies. Molecular phylogenetic analyses are generally based on 16S rRNA gene sequences in DNA extracted from the environment. The ribosome is involved in translation of messenger RNA into proteins and is therefore a universal component of all living cells. The 16S rRNA gene has ~1500 base pairs, the sequence of which has remained sufficiently conserved throughout 3.5 billion years of prokaryotic evolution to provide information on evolutionary ancestry and relationship. Based on rRNA, life is divided into the three domains of Bacteria, Archaea, and Eukarya (Woese et al., 1990). Analysis of 16S rRNA sequences provides an excellent possibility to characterize mixed microbial communities in natural environments without cultivation. The results have shown that prokaryotic diversity is much greater than indicated by previous results from half a century of cultivation and isolation. The results have also revealed numerous entirely new and so-far uncultivated phylogenetic lineages of prokaryotes (e.g., Barns et al., 1996; Hugenholtz et al., 1998).
Close phylogenic relationship of novel 16S rRNA sequences to those of cultured microorganisms with known physiologies may in many cases provide an indication of the physiology of the uncultured prokaryotes. In other cases, however, such conclusions about metabolic function may be wrong and very misleading. It is therefore important to also search for gene sequences that encode for key enzymes of metabolic pathways. Such functional genes provide information on the physiological types of microorganisms present in subsurface sediments of different geochemical zones. By comparison with 16S rRNA data the molecular ecologist can thus combine information on the identity and function of complex microbial communities. Among the important key genes analyzed from Leg 201 samples are dissimilatory (bi)sulfite reductase (dsrAB) and adenosine-5´-phosphosulfate (APS) of sulfate-reducing bacteria (Klein et al., 2001; Stahl et al., 2002) and mcrA of methanogenic archaea (Springer et al., 1995). Thus, Schippers and Neretin (in press) found the dsrA gene to be abundant in sediments of the Peru margin with 106–108 gene copies per cm3 in near-surface sediments and much lower numbers in the deep sediments. The depth gradients were steeper for the gene copy numbers than for numbers of total prokaryotes (AODC counts), which reflects the ongoing degradation of the high-molecular-weight DNA with sediment age and depth.
Results of sequencing surveys from Leg 201 samples have been reviewed by
Teske (this volume), and only some selected aspects will be discussed here. Extensive 16S rRNA clone libraries are now available for open-ocean Site 1225, Peru shelf Site 1227, Peru Trench gas hydrate Site 1230, and Peru Basin Site 1231. These libraries provide rich information on the phylogenetic diversity of the deep subsurface biosphere in this part of the ocean. It was a striking observation that the 16S rRNA clone libraries are dominated by a large number of uncultured lineages of bacteria and archaea for which nothing or very little can be deduced in terms of their basic physiology and metabolism. For other lineages, the more or less close phylogenetic affiliations with cultured organisms of known physiology motivate conclusions about the metabolic function of the unknown organisms. Their presence in different geochemical zones, which offer different potential redox couples of organic and inorganic compounds to maintain energy metabolism, appears to be an important key to their functional role in the subsurface. Yet, the distribution of specific phylogenetic groups within different biogeochemical zones dominated by sulfate reduction, manganese reduction, methanogenesis, or anaerobic methane oxidation does not provide a pattern that can be interpreted well today. Surprisingly, 16S rRNA genes representing known groups of sulfate-reducing bacteria of -Proteobacteria or known lineages of methanogenic archaea are sparse in the clone libraries, even in sediment horizons where sulfate reduction or methanogenesis should dominate microbial processes according to pore water chemistry (Parkes et al., 2005). At Site 1230, Inagaki et al. (2006) found -Proteobacteria to be more frequent in deep sulfate-free sediment than in the near-surface sulfate zone. The key gene of methanogenesis (mcrA) related to known groups of methanogens was detected in a number of samples, but their occurrence does not clearly mirror horizons with intensive methanogenesis (Parkes et al., 2005; Inagaki et al., 2006). Sequence information that couples the 16S rRNA gene with other functional genes specific for certain metabolic pathways will be needed in order to determine the relationship between prokaryotic diversity and function. Such coupled genomic information is still missing.
In accordance with this limited possibility of functional interpretation, even for major phylogenetic lineages, the new groups have been given names that mostly reflect the environment of original discovery. Among the archaea, the Deep-Sea Archaeal Group (DSAG) was originally found at hydrothermal vent sites by Takai and Horikoshi (1999) and is widely represented in the deep subseafloor, including Leg 201 sites. As a notable exception, Site 1227 is dominated by other archaeal sequences currently positioned among the Miscellaneous Crenarchaeotal Group (MCG) and the South African Goldmine Euryarchaeotal Group (SAGMEG) (Inagaki et al., 2006). At Site 1229 the MCG totally dominates the clone library analyzed by Parkes et al. (2005) and Webster et al. (in press). They found only one sequence, within the methane zone of the sediment column, that could be related to methanogens. Although such differences in archaeal dominance are interesting, there is presently no consistent explanation for the observations.
Among the bacterial sequences, uncultured members of Planctomycetes and Chloroflexi are frequently encountered (Inagaki et al., 2006). At Site 1229, sequences of green nonsulfur bacteria (GNS) and -Proteobacteria are particularly dominant (Parkes et al., 2005). Webster et al. (2004) developed 16S rDNA targeted primers for PCR to search for a novel group of uncultured bacteria that had been detected repeatedly in samples from surface and deep marine sediments. They identified a deeply branching, monophyletic cluster, designated candidate division JS-1, named in recognition of the Japan Sea as the first reported source of these sequences (Rochelle et al., 1994). The group has no cultivated relatives and its physiology is unknown, apart from its expectedly anaerobic nature based on its consistent occurrence in anoxic sediments, including Peru margin Sites 1228 and 1229 at depths to 86 mbsf. The frequency of the different bacterial groups in deep subsurface clone libraries shows some consistencies with other oceanic regions. In deep sediments from the Sea of Okhotsk, -Proteobacteria were observed to dominate in some layers whereas GNS and JS1 bacteria dominate in others (Inagaki et al., 2003).
The above examples from Leg 201 show that it is possible to extract DNA from deep sediment cores and use this DNA to sequence 16S rRNA genes that can be aligned and positioned in the phylogenetic tree of modern bacteria or archaea. The presence of intact gene sequences is, however, not in itself a proof of viable cells but could represent fossil remains of prokaryotic organisms that lived during earlier geologic periods. Thus, Inagaki et al. (2001) observed that sequences related to thermophilic and halophilic archaea are the predominant components in Pleistocene subseafloor pelagic clays. They interpreted this as remnants of a community that was trapped in the sediments at a time when the site was surrounded by hydrothermal and geothermal environments. The organisms had thus been preserved since the time of deposition under cold conditions that might not have enabled growth. It should be noted, however, that although the sequences cluster with known modern thermophiles this does not exclude the possibility that the sequences originate from organisms able to live in the temperature regime prevailing in the sediment today.
In support of the preservation hypothesis, Inagaki et al. (2005) analyzed 16S rRNA sequence diversity in and around a marine black shale horizon of ~100 Ma age, originally deposited in the ocean during a major anoxic event and now situated in southern France. Within the shale, -Proteobacteria sequences related to sulfate-reducing bacteria predominated, whereas sequences of -Proteobacteria dominate above and below the black shale. The recovered DNA signatures are consistent with the interpretation that the sequences are derived from past microbial communities buried in the subseafloor environment and preserved until the present. This has important potential implications. If such ancient sedimentary deposits can retain their genetic signals for many millions of years, then it may be possible to use such information to infer past geological conditions and microbial activities. Inagaki et al. (2005) coined the term "Paleome" for such a genetic record of past microbial communities.
Results obtained by Sørensen et al. (2004) from Site 1231 in the Peru Basin appear to contrast with this interpretation. In their study of the archaeal diversity at different subsurface depths they found at 1.8 mbsf abundant sequences of Marine Group I (MGI), members of which are highly abundant in prokaryotic picoplankton of the deep ocean. However, in deeper samples from 9 and 43 mbsf (early Pleistocene and late Oligocene age, respectively) sequences of this group were absent. These observations do not support the hypothesis of long-term survival of gene sequences from archaea buried during earlier geological history of the seafloor. Still, such conclusions are based on limited sites and data and may change as more information becomes available. The DNA of various prokaryotic groups may differ in long-term preservation as a function of the chemical and physical environment, thereby changing the pattern of extant diversity over geologic time.
Although deep subseafloor sediments may harbor more than half of all prokaryotic cells on Earth (Whitman et al., 1998), it has been unknown to what extent these organisms are alive and metabolically active. The observation of this great cell number is based on direct microscopic enumeration of bacterium-like cells containing nucleic acids that stain with fluorescent stains acridine orange or 4´,6-diamidino-2-phenylindole (DAPI). Such stains bind nonspecifically to DNA or RNA and do not provide information on the viability of the cells. Although the cells maintain morphological and macromolecular integrity, they might still be dormant or even dead (Luna et al., 2002). The presence of ribosomes in the cells, however, appears to be coupled only to metabolically active organisms. Cell death and even starvation in pure cultures is invariably associated with loss of ribosome content (e.g., Davis et al., 1986). As an indicator of living cells in Leg 201 samples, Schippers et al. (2005) therefore used a highly sensitive molecular technique targeting specifically rRNA. Using this catalyzed reporter deposition (CARD)-FISH technique, only cells with multiple ribosome copies become visible under the fluorescence microscope. The results using general probes for either bacteria or archaea show for the first time the fraction of the total cell counts that could be identified as living prokaryotes. In multiple sediment samples taken from depths as great as 427 mbsf with stratigraphic ages of up to 16 Ma, viable bacterial cells were detected. At open-ocean Sites 1225 and 1226, about one-third of the AODC-counted cells was detected by CARD-FISH analysis and should thus be alive; at ocean-margin Sites 1227 and 1230, about one-tenth was detected. It is important to note that not all living bacteria may be detected by CARD-FISH because of low ribosome content or inefficient penetration of the molecular probe into the cells. Consequently, these numbers represent minimum estimates. At all sites the abundance of archaea is apparently too low to be quantified using CARD-FISH. These observations are supported by independent quantification of 16S rDNA using quantitative, real-time polymerase chain reaction (Q-PCR) which also shows a dominance of bacteria among the prokaryotic cells (Schippers and Neretin, in press). The lack of detectable archaeal cells is in contrast to the abundance of archaea among the 16S rRNA clone libraries obtained for the same sites (Sørensen et al., 2004; Inagaki et al., 2006; Parkes et al., 2005) or the abundance of archaeal lipids detected by biomarker analyses (Biddle et al., 2006). It is too early to conclude whether the difference reflects a bias in the molecular probe technique of CARD-FISH, a bias in the extraction and analysis methods for archaeal DNA, or a bias in the conservation of archaeal versus bacterial biomarkers.
Activity Levels of Subseafloor Prokaryotes
When total cell numbers of subseafloor communities are compared to modeled rates of biogeochemical processes, such as bacterial sulfate reduction, it is obvious that metabolic rates in the deep biosphere are orders of magnitude lower than those in surface sediments (D'Hondt et al., 2002). A general conclusion from this observation could be that most microorganisms in subseafloor sediments are either inactive or adapted to extraordinarily low metabolic activity. Schippers et al. (2005) compared CARD-FISH counts of viable cells with gross sulfate reduction rates measured by 35S-radiotracer technique to calculate sulfate reduction per living bacterial cell at four sites ranging from open-ocean to ocean-margin sediments. By making some general assumptions about the energy requirement for maintenance and the potential growth yield, they calculated that turnover times of bacteria in the upper sulfate zone are in the range of 0.2 to 2 yr, both for the open-ocean and the ocean-margin sites. A similar calculation based on global estimates of carbon flux available for the subsurface bacterial community yielded turnover times of 7 to 22 yr. These values are not vastly different from turnover times of prokaryotes in soil and marine surface sediments and are considerably lower than the ~1000 yr suggested by Whitman et al. (1998) for the turnover time of the total prokaryotic population in subsurface sediments. Given this large span of calculated turnover times, we used a different approach to calculate cell-specific metabolic rate in subsurface sediments of Leg 201 sites. The results, presented in the following paragraph, also provide considerably longer turnover times.
Based on a simple transport-reaction model, D'Hondt et al. (2004) calculated for Site 1226 an areal sulfate reduction rate for the subseafloor sediment column (1.5–420 mbsf) of 1.4 x 10–7 mol/cm2/yr. Given this rate for the sediment column, the mean sulfate reduction rate per sediment volume is ([1.4 x 10–7]/[4.2 x 104] =) 3.3 x 10–12 mol/cm3/yr or 3.3 pmol/cm3/yr. Based on AODC counts of prokaryotic cells, the mean cell concentration in the upper 200 m is 5 x 106 cells/cm3. A fraction of these cells are sulfate-reducing microorganisms. Because sulfate reduction in this sediment section appears to be the predominant pathway of organic matter mineralization, we tentatively assume that 10% of all cells are sulfate reducers, similar to what was found in Arctic sediments by Ravenschlag et al. (2000). Thus, the total concentration of sulfate-reducing microorganisms, active or not, is on the order of 5 x 105 cells/cm3. The mean cell-specific sulfate reduction rate is then ([3.3 x 10–12]/[5 x 105] =) 0.0066 x 10–15 mol/cell/yr or 0.007 fmol/cell/yr. For comparison, D'Hondt et al. (2002) calculated cell-specific sulfate reduction rates for ODP ocean-margin sites of the Japan Sea, the Peru margin, and the Nankai Trough of 0.031, 0.00014, and 3.3 fmol/cell/yr, respectively, using the total cell numbers for the calculation. Had they assumed that only 10% are sulfate reducers, they would have calculated 10-fold higher cell-specific rates.
Cell-specific sulfate reduction rates in surface sediments have been calculated for a small number of coastal marine ecosystems from direct quantification of the sulfate-reducing populations using RNA- and DNA-based techniques and from direct measurements of sulfate reduction rates using 35S-labeled sulfate. In temperate coastal sediments, Sahm et al. (1999) calculated rates of 3–30 fmol/cell/yr, whereas in Arctic sediments of Svalbard, Ravenschlag et al. (2000) calculated rates of 10–50 fmol/cell/yr, ~1000-fold higher than cell-specific rates calculated for Site 1226 subsurface sediments. In comparison, cell-specific rates for sulfate-reducing bacteria in laboratory cultures tend to be much higher, for example, 300–1000 fmol/cell/yr (mean = 500 fmol/cell/yr) at 0°C for cold-adapted sulfate reducers isolated from Arctic sediments (Knoblauch et al., 1999). This is 40,000-fold higher than the cell-specific rates at Site 1226. With a growth yield of 3–7 g dry weight of biomass per mole of organic substrate consumed, these pure cultures of sulfate reducers typically have doubling times of 3–30 days (mean = 10 days) (Knoblauch and Jørgensen, 1999). At a given growth yield, there is an inverse relationship between cell-specific metabolism and doubling time. Thus, if we assume that sulfate-reducing microorganisms of Site 1226 have a similar growth yield as typical pure cultures, then their mean turnover time would be 40,000-fold longer (i.e., 400,000 days or ~1000 yr). In reality, the growth yield in the deep subsurface is expectedly lower than in pure culture, given the extremely slow growth and the proportionally larger energy requirement for maintenance of cell functions. The turnover time would therefore be correspondingly longer than 1000 yr. Recently, Biddle et al. (2006) calculated turnover times for subsurface prokaryotic communities in Leg 201 Peru margin cores of 100–2000 years.
Although these calculations yield a large span of potential turnover times, the extremely low growth rates of deep biosphere microorganisms remains enigmatic and without comparison to the much faster growing laboratory cultures. One explanation for the large difference in mean growth rates may still be that only a fraction of the bacterial population is actively growing while a large fraction may be inactive at a given time. There is accumulating evidence, both from ocean water and from sediments, that only some of the prokaryotic cells that can be quantified by direct epifluorescent counting (AODC) are metabolically active and contribute to microbially catalyzed processes (e.g., Luna et al., 2002).
It remains a challenging question in deep biosphere research how cells can maintain molecular integrity and metabolic function under the extreme low energy conditions offered by their environment, whether their turnover time is a few years or many thousand years. As a deposited sediment layer becomes buried deeper and deeper below the seafloor over millions of years, the residual organic matter in this layer becomes increasingly refractory. A mass balance calculation of the progressively slower degradation vs. the lapsed time since initial deposition indeed shows that the available energy flux from buried organic carbon is extremely low. The gradual heating during burial due to the geothermal gradient may stimulate the degradability of the organic matter, although it obviously does not increase its total pool size. Wellsbury et al. (1997) suggested that deep geothermal heating in sediments at Blake Ridge, northwest Atlantic Ocean, could explain an observed increase in pore water acetate concentrations of 2–3 orders of magnitude relative to surface concentrations. A similar release of acetate was demonstrated experimentally by heating coastal surface sediments and could provide a substrate for sulfate-reducing or methanogenic microorganisms. Although this mechanism is well established for deep oil reservoirs at temperatures >80°C (Cooles et al., 1987; Borgund and Barth, 1994) and was experimentally induced at much lower temperatures (Wellsbury et al., 1997), the presence of high acetate concentrations at Blake Ridge is surprising. The in situ temperatures at 700 mbsf at Blake Ridge do not exceed 30°C, which is well within the temperature range of mesophilic prokaryotes. The high acetate concentrations therefore indicate that if the acetate is indeed utilized by microorganisms, its turnover remains extremely slow and appears to be limited by factors other than acetate availability. Otherwise, the microorganisms should be able to consume this widely used energy source of sulfate reducers and methanogens within a relatively short period.
Chemical Interfaces: Case Study of Site 1229
Chemical interfaces in the subsurface exist where diffusible species meet and are consumed in energy-yielding redox processes. Such processes may be used for energy metabolism of specialized prokaryotes and thereby provide zones of enhanced metabolic activity and increased population density. Such an enhancement is well known from chemical interfaces in more shallow sediments, for example, at the transition between sulfate and methane. A specialized microbial community of sulfate-reducing bacteria and methane-oxidizing archaea grows where sulfate and methane coexist. The two groups of prokaryotes may form unique consortia consisting of aggregates with a central colony of archaea and a peripheral coating of sulfate reducers (Boetius et al., 2000; Orphan et al., 2001). It is assumed that the archaea transfer reducing equivalents from methane to the sulfate reducers, for example in the form of H2, but the nature of this transfer is still not clear. The energy yield of the reaction available for organisms is marginal, ~20 kJ/mol CH4, and it even has to be shared among the two partners in the consortium. In recent years, a large diversity of methane-oxidizing archaea has been identified in many different sediment environments (Knittel et al., 2005). Some of the archaea do not form consortia with sulfate reducers but appear to grow as individual cells in the sulfate–methane transition zone. It is an interesting question whether such archaea possess the entire enzymatic machinery to catalyze both the methane oxidation and the sulfate reduction within a single cell, and thereby more than double the energy yield for the individual cell.
Parkes et al. (2005) analyzed the sulfate–methane transitions at organic-rich Peru margin Site 1229. This site is unusual as it has a deep brine incursion, so sulfate penetrates both downward from the overlying seawater and upward from the underlying brine. Between these two sulfate sources, methane accumulates to ~2 mM in the intermediate sulfate-free zone and meets the sulfate by diffusion into transition zones at ~30 and 90 mbsf (Shipboard Scientific Party, 2003c) (Fig. F5A). There is a striking increase in cell densities at the two sulfate–methane transitions, 10- to 1000-fold above those in the sediment above and below the transitions (Fig. F5B). At 90 mbsf, where the sediment age is ~0.8 Ma, the highest cell densities ever encountered in subsurface sediments were detected, 1 x 1010 cells/mL sediment. This density even exceeds by 10-fold that in the surface sediment at Site 1229. The distinct peaks in population size occur exactly at the two sulfate/methane diffusion interfaces where there is enhanced energy supply. This zonation shows that, at least in some cases, the microbial community does indeed respond to the modern chemical environment.
Sulfate reduction was measured experimentally in samples from Site 1229 and showed an interesting distribution (Fig. F5C). Highest reduction rates were recorded near the sediment surface and in the uppermost 10 m of sediment. Below 10 mbsf rates were mostly below detection limit and only the data points shown in Figure F5C were significantly above detection. On that background, peaks in reduction occur near the two sulfate–methane transitions, presumably fueled by anaerobic methane oxidation. The absolute rates of sulfate reduction (<10 pmol SO42–/cm3/d) are similar to the measured rates of methanogenesis. The rates are also extremely low, 2–3 orders of magnitude lower than rates normally recorded in coastal sediments, and are near the theoretical limit of detectability for 35S radiotracer. Although the absolute rates of sulfate reduction are therefore not accurate, the distribution of activity still provides a clear picture of where the hotspots are found today in the deep subsurface. In spite of ongoing sulfate reduction, sulfate-reducing bacteria were not detected in the sequence libraries, either near the sediment surface or at the two sulfate–methane transitions (Parkes et al., 2005). The dsr gene, specific for sulfate-reducing bacteria, could also not be detected among the PCR products, which seems to confirm a low representation of the known types of sulfate reducers in the community.
Analyses of 16S rRNA gene diversity throughout the sediment, using denaturing gradient gel electrophoresis (DGGE) and subsequent sequencing of visible bands (Parkes et al., 2005; Fry et al., in press), show that the microbial community at 90 mbsf differs distinctly from that in the rest of the sediment column. Although methanogens were generally absent in the 16S rRNA clone libraries, the methanogen-specific gene, mcrA, was found at all depths analyzed, with sequence relationship to the Methanobacteriales and Methanosarcinales. Members of these groups utilize H2/CO2 and/or acetate for methane formation, which is consistent with the measured methane formation from both these substrates (Fig. F5D). The bacteria in the deep subsurface are thus not just survivors of earlier populations that grew at the sediment surface at a time when the energy supply was higher but have adjusted to the present chemical zonation. Site 1229 is an excellent example of such a bacterial adaptation to the chemical environment because the zonation pattern occurs twice within a sediment column covering the entire Pleistocene.
Figure F5A shows how the zones of sulfate and methane are separated, as the presence of sulfate prevents the accumulation of methane. The zones of sulfate reduction and methanogenesis are generally separated in marine sediments. Experiments have shown that sulfate reducers can outcompete methanogens for common, limiting substrates and thereby suppress methane formation. The two processes are, however, not completely mutually exclusive because methanogens can use small quantities of noncompetitive substrates and because the competition is not totally efficient. Parkes et al. (2005) used a highly sensitive radiotracer technique to measure the rate of methanogenesis in the Site 1229 sediment column. They found that methane production does in fact take place in the sulfate zone and that methane is preferentially formed from CO2 and H2 in the upper sediment layers and from acetate deeper in the core with rates up to 15 pmol/cm3/d (Fig. F5D). Methane does not accumulate in the sulfate zones, however, possibly because it is being removed by anaerobic methane oxidizers. It is not clear how concurrent methane formation and oxidation is energetically possible because, from a simplistic point of view, a process can be exergonic only in one direction.
Biological formation of methane within sulfate-rich sediment was also observed in sediments of the open Pacific sites (D'Hondt, Jørgensen, Miller, et al., 2003) and sites throughout the world ocean (D'Hondt et al., 2002). Concentrations of methane are very low and reflect the correspondingly low levels of organic turnover. Yet, the occurrence of methane within the sulfate zone demonstrated that methanogenesis and sulfate reduction are not mutually exclusive, even in sediments where the competition for the scarce energy resources must be fierce.
