GEOCHEMISTRY
The geochemistry program included characterization of volatile gases, interstitial water, sedimentary inorganic carbon, organic carbon, nitrogen, and sulfur, and organic matter type and maturity. These analyses were carried out as part of the routine shipboard safety and pollution prevention requirements and to provide preliminary information for shore-based research.
Sediment Gases Sampling and Analysis
During Leg 202, the compositions and concentrations of volatile hydrocarbons and other gases in the sediments were monitored at typical intervals of one per core. Samples were obtained by two different methods. The routine headspace procedure (Pimmel and Claypool, 2001) involved placing approximately 5 cm3 of sediment sample in a 21.5-cm3 glass serum vial that was sealed with a septum and metal crimp cap and heated at 60°C for 30 min. A 5-cm3 volume of gas from the headspace in the vial was removed with a glass syringe for analysis by gas chromatography.
A second gas sampling procedure, the vacutainer method, was used for gas pockets or expansion voids that appeared in the core while it was still in the core liner. A device with a heavy-duty needle was used to penetrate the core liner, and an attached syringe was employed to collect the gas.
Headspace and vacutainer gas samples were both analyzed using a Hewlett-Packard 5890 II Plus gas chromatograph (GC) equipped with a 2.4 m x 3.2 mm stainless steel column packed with 80/100 mesh HayeSep S and a flame ionization detector (FID). This instrument quickly measures the concentrations of methane (C1), ethane (C2), ethene (C2=), propane (C3), and propene (C3=). The gas syringe was directly connected to the GC via a 1-cm3 sample loop. Helium was used as the carrier gas, and the GC oven temperature was held at 90°C. Data were collected and evaluated with a Hewlett-Packard 3365 Chemstation data-handling program. Calibrations were done using Scotty IV analyzed gases, and gas concentrations were measured in parts per million by volume.
When high concentrations of C2+ hydrocarbons or of nonhydrocarbon gases such as H2S or CO2 were suspected, gas samples were analyzed with the Natural Gas Analyzer (NGA), which routinely measures hydrocarbons through C6. The NGA system consists of a Hewlett-Packard 5890 II Plus GC equipped with multiport valves that access two different column and detector combinations. Hydrocarbons from methane to hexane were measured with a 60 m x 0.32 mm DB-1 capillary column and an FID. The GC oven holding this column was heated from 80° to 100°C at 8°C/min and then to 200°C at 30°C/min. Nonhydrocarbon gases were isothermally analyzed at the same time using a sequence of packed columns (15-cm HayeSep R column connected to a 1-m molecular sieve column and a 2-m Poropak T column) and thermal conductivity detectors (TCDs). Helium was the carrier gas in both systems, and a Hewlett-Packard Chemstation was used for data acquisition and processing. Chromatographic response was calibrated against preanalyzed standards. Gas contents are reported in parts per million by volume.
Interstitial Water Sampling and Chemistry
Interstitial waters were extracted from 5- to 10-cm-long whole-round sections that were cut and capped immediately after core retrieval on deck. In one hole at each site, samples were taken from each core for the upper 100 mbsf and, depending on the site and depth range, at intervals from every core to every second to third core thereafter to total depth. Occasionally, samples from more than one hole were treated as constituting a single depth profile using mcd as the depth reference. At Sites 1239 and 1240, interstitial water samples were taken more frequently (one per section for the upper 60 mbsf) for shore-based analyses of oxygen isotopes, deuterium, and chlorinity at high precision. Before squeezing, samples were removed from the core liner and the outside surfaces were carefully scraped off with spatulas to minimize potential contamination. Whole rounds were placed into a titanium and steel squeezing device and squeezed at ambient temperature with a hydraulic press. Interstitial water samples were collected in plastic syringes, filtered through 0.45-µm Gelman polysulfone disposable filters, and stored in plastic sample tubes for shipboard analyses or archived in glass ampoules and/or heat-sealed acid-washed plastic tubes for shore-based analysis.
Interstitial water analyses followed the procedures outlined by Gieskes et al. (1991) and Murray et al. (2000) with modifications as indicated. Interstitial water samples were analyzed for salinity with a handheld refractometer; for pH and alkalinity by Gran titration with a Brinkman pH electrode and Metrohm autotitrator; for Cl- concentrations by titration; for SO42- concentrations by ion chromatography with a Dionex DX-120 ion chromatograph; for H4SiO4, HPO42-, and NH4+ concentrations by spectrophotometric methods with a Milton Roy Spectronic 301 spectrophotometer; for Na+, Mg2+, Ca2+, and K+ by ion chromatograph (Site 1232) or by inductively coupled plasma-atomic emission spectroscopy (ICP-AES) with a Jobin Yvon JY2000; and for Mn2+, Fe2+, B, Sr2+, Ba2+, and Li+ by ICP-AES. Sample and standard aliquots used for HPO42- determinations were acidified with 1-N HCl to a pH <2 to avoid analytical artifacts related to the possible presence of hydrogen sulfide in interstitial water samples. ICP-AES techniques for the major cations Na+, Mg2+, Ca2+, and K+ used dilutions of International Association of Physical Sciences Organization (IAPSO) standard seawater as calibration standards. Standards and samples were diluted 1:5 with distilled water then 1:10 with a 2.5% HNO3 (by volume) matrix solution with yttrium at 10 ppm as an internal standard. Because of the matrix influence of variable Na+ in IAPSO dilutions on K+ intensity, K+ calibration curves were constructed using only a blank and the 100% IAPSO dilution. ICP-AES techniques for the minor elements Mn2+, Fe2+, B, Sr2+, Ba2+, and Li+ were modified from those described by Murray et al. (2000) by preparing calibration standards in an acidified (2.5% HNO3, by volume) sodium chloride matrix (35 g NaCl/L) and by using the 2.5% HNO3 matrix solution with yttrium at 10 ppm as an internal standard to dilute standards and acidified interstitial water samples 1:10 for each analytical run.
IAPSO standard seawater was used for calibrating techniques when applicable. The accuracy of techniques was assessed by determining IAPSO or solutions of known composition as samples and calculating the measured/expected value as a ratio. The reproducibility of techniques, expressed as 1-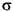 relative standard deviations of means, was evaluated by replicate analyses of drift solutions similar in composition to samples, of IAPSO treated as a sample, and/or of samples both within a given analytical run and in different analytical runs (Table T7). Na+ was also determined by charge balance, neglecting contributions by ammonium and bromide, with the charge balance values reported here. Chemical data for interstitial water are reported in molar concentration units in each site report.
relative standard deviations of means, was evaluated by replicate analyses of drift solutions similar in composition to samples, of IAPSO treated as a sample, and/or of samples both within a given analytical run and in different analytical runs (Table T7). Na+ was also determined by charge balance, neglecting contributions by ammonium and bromide, with the charge balance values reported here. Chemical data for interstitial water are reported in molar concentration units in each site report.
Sedimentary Inorganic Carbon and Organic Carbon, Nitrogen, and Sulfur Concentrations
Inorganic carbon concentrations were determined using a Coulometrics 5011 carbon dioxide coulometer equipped with a System 140 carbonate analyzer. One carbonate determination was performed typically for each 1.5-m section of core. Samples of ~15 mg of freeze-dried, ground sediment were reacted with 2-N HCl. The liberated CO2 was backtitrated to a colorimetric end point. Calcium carbonate content, as weight percent, was calculated from the inorganic carbon (IC) content with the assumption that all inorganic carbon is present as calcium carbonate:
-
% CaCO3 = % IC x 8.33.
Ten replicate analyses of aliquots from a high calcium carbonate sample (202-1236A-3H-3, 74-75 cm) range from 95.5 to 96.1 wt% (mean = 95.8 wt%; standard deviation = 0.2 wt%) (Table T8).
Total carbon, total nitrogen, and total sulfur were determined using a Carlo Erba 1500 CNS Analyzer on a subset of the samples used for inorganic carbon determinations. Aliquots of 10 mg of freeze-dried, ground sediment with ~10 mg V2O5 oxidant were combusted at 1000°C in a stream of oxygen. Nitrogen oxides were reduced to N2, and the mixture of N2, CO2, H2O, and SO2 gases was separated by GC and detection performed by TCD. The H2 values are not useful because they represent hydrogen from both organic matter (OM) and (clay) minerals. All measurements were calibrated by comparison to pure sulfanilamide as standard. Contents of TOC, as weight percent, were calculated as the difference between total carbon (TC) and inorganic carbon (IC):
-
% TOC = % TC - % IC.
No formal assessment of analytical precision for organic carbon determinations by difference was made (e.g., by multiple measurements of a single sample). An earlier comparison of total organic carbon determinations by difference made shipboard with shore-based analysis of the same samples on the carbonate-free fraction found that shipboard organic carbon determinations for Iberia Abyssal Plain sediments (total organic carbon typically up to 1 wt%, some as high as 2.5 wt%) were systematically low relative to shore-based measurements (Meyers and Silliman, 1996). One-third of the samples agreed within 0.1 wt%, but differences ranged from <0.1 wt% to 0.5 wt% and occasionally much greater (1 wt% or more). Assuming that typical absolute error on calcium carbonate determinations is ±0.2 wt% (see Table T8) (equivalent to an absolute error of about ±0.02 wt% for inorganic carbon), estimates of possible errors in total carbon determinations allow the estimation of errors in total organic carbon. For example, a sample with 75 wt% calcium carbonate and 1 wt% total organic carbon would have absolute errors in total organic carbon of ±0.03-0.10 wt% if total carbon determinations had relative errors of ±0.02%-1.0% relative standard deviation. For a sample with 8.3 wt% calcium carbonate and 1.0 wt% total organic carbon, absolute errors would be ±0.02-0.03 wt% for the same range of relative errors in total carbon determinations. The errors in total carbon are not defined, so these estimates of errors in total organic carbon by difference should be used only as a guide.
Organic matter atomic carbon/nitrogen ratios were calculated from TOC and total nitrogen concentrations to characterize the nature of the organic matter (i.e., to distinguish between marine and terrigenous sources) (Meyers, 1997).
The type of organic matter was characterized in a selected set of samples (organic carbon-rich sediments) by pyrolysis using a Delma Nermag Rock-Eval II pyrolysis system. This method is based on a whole-rock pyrolysis technique to identify the type and the maturity of the organic matter and to detect the petroleum potential of the sediments (Espitalié et al., 1986). The Rock-Eval system includes a temperature program that first releases volatile hydrocarbons (S1 peak) at 300°C for 3 min. Then, hydrocarbons are released through thermal cracking of kerogens (S2 peak) as the temperature is increased from 300° to 550°C at 25°C/min. S1 and S2 parameters are measured with an FID and are reported in milligrams per gram of sediment. The temperature at which the kerogen yields the maximum amount of hydrocarbons during the S2 program provides Tmax, a parameter that indicates the maturity of the organic matter. Between 300° and 390°C of the pyrolysis program, CO2 released from the thermal degradation of organic matter (S3 peak) is trapped and measured by TCD in milligrams per gram of sediment. Rock-Eval II parameters characterize organic matter by allowing the hydrogen index (HI; in milligrams of hydrogen per gram of organic carbon) to be calculated:
-
HI = S2 x 100/TOC.
Interpretation of Rock-Eval data is considered to be unreliable for samples containing <0.5 wt% TOC (Peters, 1986).
