RESULTS AND DISCUSSION
Hydrogen and Oxygen Isotopic Compositions
Water isotopic compositions from Leg 204 are listed in Table T1 and shown in Figure F2, which includes data from Site 888 located west of the deformation front of Cascadia margin (Kastner and Elderfield, 1995). Similar to the approach used by Tréhu et al. (2004) to estimate in situ Cl– values, the background levels of 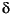 D and 18O can be defined by interpolating values from the interstitial waters collected from sediments devoid of gas hydrates. (Fig. F3). The chemistry of such water is assumed to be free from the effects of gas hydrate formation and dissociation, and thus should reflect in situ values.
D and 18O can be defined by interpolating values from the interstitial waters collected from sediments devoid of gas hydrates. (Fig. F3). The chemistry of such water is assumed to be free from the effects of gas hydrate formation and dissociation, and thus should reflect in situ values.
The D values at the summit and flank sites increase from –5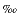 near the seafloor to ~5 at ~20 mbsf, show a very slight decrease downward, and then remain almost constant at values ranging from 0 to +4 (Fig. F2B). On the other hand, D values in the slope basin decrease steeply downward, reaching values of about –10, and remain almost constant below 220 to 300 mbsf, with maximum values as high as 5 at ~20 mbsf. Positive (up to +20) and negative (up to –10) excursions in D at all sites always accompany depletion and enrichment in Cl– (Fig. F2A), respectively.
near the seafloor to ~5 at ~20 mbsf, show a very slight decrease downward, and then remain almost constant at values ranging from 0 to +4 (Fig. F2B). On the other hand, D values in the slope basin decrease steeply downward, reaching values of about –10, and remain almost constant below 220 to 300 mbsf, with maximum values as high as 5 at ~20 mbsf. Positive (up to +20) and negative (up to –10) excursions in D at all sites always accompany depletion and enrichment in Cl– (Fig. F2A), respectively.
The 18O values increase from 0 near the seafloor to +0.5 at ~20 mbsf at all sites, and then decrease downward, approaching approximately –0.3 below 100 mbsf at the summit and flank sites, whereas they gradually decrease to values as low as –0.8 and become constant below 220 to 300 mbsf in the slope basin (Fig. F2C). Positive (up to +2.4) and negative (up to –0.9) excursions in 18O at all sites correspond with D excursions and with anomalous depletion and enrichment in Cl–, respectively.
Anomalies Associated with Gas Hydrate
The "spiky" decreases of dissolved Cl– observed within the GHSZ (e.g., Holes 1244C, 1245B, and 1251D in Fig. F3) are accompanied by increases in D and 18O. These discrete anomalies are clearly associated with gas hydrate dissociation during core retrieval. Because the gas hydrate lattice excludes dissolved ions and preferentially incorporates D and 18O during formation, the interstitial water released from gas hydrate as it dissociates shows low Cl– concentration and high D and 18O values in direct proportion to hydrate content (Egeberg and Dickens, 1999; Hesse et al., 2000; Matsumoto and Borowski, 2000). Conversely, strong enrichment in Cl– in shallow depths at Site 1249 at the ridge summit may reflect rapid gas hydrate formation, which exceeds the rate at which ions can be removed by diffusion from the loci of hydrate formation (Torres et al., 2004a) or great reduction of sediment pore space due to massive gas hydrate formation, which leads to decrease of dissuasive loss of Cl– (Milkov et al., 2004). The briny residual waters show negative excursions in D and 18O because the water is depleted in D and 18O because of isotopic fractionation during gas hydrate formation.
Glacial Signals in the Shallow Interstitial Waters
Sites away from the ridge crest show curvature in Cl–, D, and 18O in shallow sediments (e.g., Sites 1244, 1251, and 1252 as shown in the boxes in Fig. F4). Small positive peaks in Cl– (Fig. F4A) in comparison with seawater are observed shallower than 40 mbsf in Holocene to Pleistocene sediments (Tréhu, Bohrmann, Rack, Torres, et al., 2003) and correspond with positive peaks in D (Fig. F4B) and 18O (Fig. F4C). The methane concentration in the shallow sediments drilled away from the ridge summit is not sufficient to form gas hydrate at these depths (Milkov et al., 2003); thus, the upper ~40 mbsf of these sites are devoid of gas hydrates, as shown by analyses of multiple gas hydrate proxies (Fig. F3) (Tréhu et al., 2004). The observed synchronous changes in dissolved Cl– and water isotopes in the upper sediments (Fig. F4) are therefore not caused by processes related to gas hydrate formation; rather, these profiles reflect the burial of seawater during glacial intervals which is enriched in dissolved ions, D, and 18O because of ice sheet development (e.g., Adkins et al., 2002).
Detailed analyses of interstitial waters recovered from the Pacific, Southern, and Atlantic oceans show a Cl– increase of ~2.6% and a 18O rise of 0.8 (McDuff, 1985; Schrag et al., 1996; Adkins et al., 2002), which has been used to constrain seawater temperature and salinity values from the Last Glacial Maximum (LGM) to the Holocene. In Cascadia continental margin sediments, dissolved Cl– shows an increase of ~3.6% (Fig. F4A) and 18O increases by ~0.7 (Fig. F4C) relative to modern seawater. Our data set provides the first D values associated with the postulated interstitial water records of ice volume change in glacial to interglacial timescales. We show a maximum increase in D of ~4 (Fig. F4B) in the upper 40 mbsf, which is about six times greater than that observed for 18O. This magnitude is consistent with the experimental results of oxygen and hydrogen isotopic fractionation for an ice-water system (O'Neil, 1968) and suggests that the observed coupled increase in D and 18O is due to the development of ice sheet resulting in the sea level drop during the LGM. The effects of older glacial-interglacial periods in deeper interstitial waters are likely overwritten by diffusion and advection of deeper fluids.
Fluid Provenance Inferences for Interstitial Water from Cl– and D Profiles
The similarity in profiles of Cl– and D (Fig. F2) suggests that same processes control the vertical distributions of these species; both Cl– and D generally decrease with depth in the slope basin, whereas those remain almost constant at the other sites. Anaerobic methane oxidation (e.g., Borowski et al., 1996),
-
CH4 + SO42–
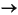 HS– + HCO3– + H2O, (1)
HS– + HCO3– + H2O, (1)
provides water depleted in D because the hydrogen in water can be linked to bacterially generated methane with D values of approximately –200 (Coleman et al., 1981; Waseda and Uchida, 2002). Therefore, this process can decrease both Cl– and D. But the methane concentration is negligibly small compared with the amount of hydrogen in water (Dählmann and de Lange, 2003), and the sulfate reduction zone in Hydrate Ridge is very shallow (generally <15 mbsf) (Tréhu, Bohrmann, Rack, Torres, et al., 2003); thus, this process cannot account for the magnitude of the observed decrease in D in deep sediments.
Membrane filtration of clay minerals causes interstitial waters to be depleted both in Cl– and D as well as 18O. Experiments have shown that Cl– concentration can decrease by 18% and 30% in water filtered through bentonite at pressures of 14 and 28 MPa, respectively (Kharaka and Berry, 1973). In addition, Coplen and Hanshaw (1973) showed that this process results in depletion of D by 2.5 and depletion of 18O by 0.8 when water is filtered through montmorillonite at 33 MPa. But membrane filtration, which has been shown experimentally to cause freshening and depletion in the heavy isotopes, has never been documented unambiguously in a large-scale field example (Hanor, 1987). In the slope basin at Hydrate Ridge, we observe decreases of 12%–23%, 5–10, and 0.5–1 from seawater values of Cl–, D, and 18O, respectively (Fig. F2). If we assume that these values are due to membrane filtration at depth followed by fluid migration to shallower sedimentary section, the expected water isotopic fractionation observed in the above experiments are too small to cause the large depletion of D and 18O in the interstitial waters.
However, water released during clay mineral diagenesis is a promising explanation for overall decreases in Cl– and D observed in the eastern slope basin of the ridge. Torres et al. (2004b) explain the progressive freshening of fluids landward as resulting from dehydration of smectite beneath the accreted mélange and subsequent migration of fluids from depths below 1000 mbsf where in situ temperature is >70°C. Hydrogen isotopic fractionation between smectite and water has been determined by Capuano (1992) using the relationship
-
1000 ln
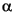 Hclay-water = {[–45.3(103)]/T} + 94.7, (2)
Hclay-water = {[–45.3(103)]/T} + 94.7, (2)
where Hclay-water is the isotopic fractionation factor of hydrogen between smectite and water and T is the fractionation temperature in degrees K. If we assume that the observed decrease in background level of Cl– is caused by input of freshwater evolved from smectite dehydration at greater depth, we can estimate average isotopic fractionation temperature between clay and water using a mass balance calculation such that
-
Xclayclay + (1 – Xclay)SW = IW, (3)
where Xclay is the volume fraction of water from clay mineral dehydration calculated from Cl– change and clay, SW, and IW represent the D values of water released by dehydration, seawater, and the predicted composition of the interstitial water, respectively. Using Equations 2 and 3, we can estimate the average temperatures of hydrogen isotopic fractionation between clay and water that fit to the observed decreasing D profiles in the slope basin. The results indicate that in situ interstitial waters were released from clay minerals isotopically exchanged at average temperatures of 28°, 52°, and 66°C at Sites 1244, 1252, and 1251, respectively, as dashed lines in Figure F4B. Using the regional geothermal gradient for these sites (Tréhu, Bohrmann, Rack, Torres, et al., 2003), these temperatures correspond to subbottom depths of 412, 825, and 1103 mbsf, respectively. These temperatures are too low for dehydration to occur (e.g., Perry and Hower, 1970; Pytte and Reynolds, 1989) and must reflect previous fractionation of interlayer water with the pore fluids at depth during burial, so we are not starting out with seawater values in our Equation 3. Nevertheless, the apparent temperature increases landward in the slope basin, consistent with our conceptual understanding of prism evolution based on models developed for the Barbados wedge (Bekins et al., 1994).
Causes of Depletion in 18O
Decreases in Cl– and D values are observed only in the eastern slope basin; 18O values, however, decrease at all sites. Clay mineral dehydration in the slope basin also affects the oxygen isotopic composition of the interstitial water. The temperature dependence of isotopic fractionation of oxygen between smectite and water was determined by Savin and Lee (1988) and is given by
-
1000 ln oclay-water = {[2.60(106)]/T2} – 4.28, (4)
where oclay-water is the isotopic fractionation factor of oxygen between smectite and water and T is the fractionation temperature in degrees K. Such clay minerals are enriched in 18O and increase 18O of interstitial water during dehydration (e.g., Dählmann and de Lange, 2003). Dashed lines in Figure F4C show the predicted initial background levels for 18O of the water isotopically exchanged with clay at temperatures of 28°, 52°, and 66°C at Sites 1244, 1252, and 1251, respectively, if it was derived by clay mineral dehydration. The observed decreases in 18O profiles do not follow the predicted trends; thus, the 18O values in the slope basin were generated by the combined processes of clay mineral dehydration and other geochemical reactions. The actual 18O depletion in the slope basin due to reactions other than clay mineral dehydration must be greater than that at the summit and flank sites; a 18O decrease of ~5 in the slope basin (Fig. F4C) and ~0.3 at the other sites (Fig. F2C) occurs because the 18O values in the slope basin were potentially increased by the clay mineral dehydration to the values indicated as the dashed lines in Figure F4C.
Carbonate precipitation is an important process that can decrease 18O of interstitial water independent of D and is described by the following general reaction:
-
M2+ + 2HCO3– MCO3 + H2O + CO2, (5)
where M is Ca2+ or Mg2+, representing the formation of calcite or dolomite. The isotopic fractionation factors (ocarbonate-water) of oxygen between carbonates and water are experimentally determined as calcite,
-
1000 ln ocalcite-water = {[2.78(106)]/T2} – 3.39, (6)
from O'Neil et al. (1969), and dolomite,
-
1000 ln odolomite-water = {[2.62(106)]/T2} + 2.17, (7)
from Fritz and Smith (1970). Assuming that carbonate precipitation occurred at 276.85 K (3.7°C) and 293.75 K (20.6°C), equivalent to depths at the seafloor and 300 mbsf at Site 1251 (Tréhu, Bohrmann, Rack, Torres, et al., 2003), the estimated isotopic fractionations are +32.9 (SMOW) at 3.7°C and +28.8 at 20.6°C for calcite and +36.4 and +32.5 for dolomite, respectively. The precipitation of carbonates accounts for the observed decreases in 18O of interstitial water at all sites (Fig. F2C). Thus, the predicted greater decrease in 18O (~5) in the slope basin, compared to that at the summit and flank sites (~0.3) as mentioned above, likely reflects abundant authigenic carbonate and possibly high dolomite content and/or low precipitation temperature. Alternatively, Sr2+ concentration and the strontium isotopic composition (87Sr/86Sr) of interstitial waters measured in these fluids suggest the influence of fluid interaction with oceanic basement (Torres et al., 2004b; Teichert et al., 2005). The 18O values of oceanic basement recovered from the eastern flank of Juan de Fuca Ridge during ODP Leg 168 range from +6.1 to +19.3 (Vienna SMOW), showing a positive correlation between 18O and the percentage of bulk rock alteration (Hunter et al., 1999). Oceanic basement in contact with seawater preferentially incorporates 18O into its alteration products, resulting in a significant decrease of 18O of interstitial waters. The effect of basalt alteration in deep fluids at Hydrate Ridge shows an increase eastward (landward) (Torres et al., 2004b), consistent with the observed greater decrease in 18O in the eastern slope basin of the ridge relative to the summit and flank regions.
