DISCUSSION
Chemical Composition of Interstitial Waters
Natural gas geochemistry at southern Hydrate Ridge is complicated by gas migrating from depth and mixing with gas generated in the shallow sediments. Gas samples with the migrated gas component were recovered in shallow sediments at Sites 1248, 1249, and 1250 and in sediments in and near Horizon A at Sites 1245 and 1247. The slope basin sites (1244, 1251, and 1252) and ridge saddle sites (1245, 1246, and 1247) at shallow depths represent sediments dominated by microbial methane generated during early diagenesis. Gas hydrate concentrations are much higher at the summit sites compared with sites away from the summit (20% of pore volume compared with 2%–5% of pore volume), indicating the importance of gas transport for sustaining higher concentrations of gas hydrate in surface sediments. Gas hydrates at nonsummit sites do not form until organic matter remineralization and microbial methanogenesis builds dissolved methane concentrations to hydrate solubility levels. Thus, there are two contrasting styles of gas hydrate formation at south Hydrate Ridge, here termed "transport-dominated" and "reaction-dominated" gas hydrate systems.
Transport-Dominated Gas Hydrate
One of the most important results of Leg 204 was determining the gas geochemistry, which defines the dominant migration pathways for gas from depth to the seafloor and results in venting of gas and formation of gas hydrate in surface sediments (Milkov et al., 2005). The pathways are localized in the summit region first by Horizon A, a northeast-dipping high-amplitude reflector shown by coring and logging to contain methane-rich gas at high gas saturation levels (40%–80%) with a significant component of C2–C5 hydrocarbons of deeper, thermogenic origin; second by the intersection of Horizon A with the base of the GHSZ; third by migration of gas through vertical gas chimneys above these intersections to the seafloor and near-surface sediments; and then laterally up to the topographic summit. These migration pathways are traced by means of the distinctive gas geochemistry, mainly the isotopically heavy C2–C5 hydrocarbons. However, the methane component of the total gas stream is isotopically light methane (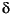 13C1; from –65
13C1; from –65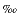 to –62) of apparent microbial origin. The methane component of the gas must be dominated by previously generated microbial methane buried with sediments of the accretionary prism that is now migrating along permeable pathways back to the seafloor, and carrying with it a minor component of thermogenic gas.
to –62) of apparent microbial origin. The methane component of the gas must be dominated by previously generated microbial methane buried with sediments of the accretionary prism that is now migrating along permeable pathways back to the seafloor, and carrying with it a minor component of thermogenic gas.
Proportions of Gas Sources
At the summit of Hydrate Ridge, the gas appears to result from three sources: (1) a minor thermogenic source characterized by a full suite of isotopically heavier C2–C5 hydrocarbons, (2) microbial methane that was buried with and then exsolved from accreted sediments, and (3) microbial methane currently being generated at shallow depths of burial. The relative proportions of these three sources can be roughly estimated. The fact that there is three or four times as much gas hydrate at the summit sites as there is at the nonsummit sites (Tréhu et al., 2004), where only normal microbial processes are operating, suggests that the exsolved and migrated microbial methane plus thermogenic component is ~75%–80% of the gas at the summit. The proportion of this migrated component made up by thermogenic gas can be estimated from the chemical and isotopic composition. The 13C of C2–C5 components can be used to infer that the 13C of thermogenic methane is about –45, according to the method outlined by Chung et al. (1988) and illustrated in Figure F7. If the end-member composition of the thermogenic component, in terms of 13C of methane and C1/C2 ratio, is –45 and 10, and for the microbial component is –68 and 10,000, then simple two-component mixing models suggest that the proportion of thermogenic gas in the migrated hydrocarbon stream is ~20%–25%. This means the relative proportions of the three source components at the summit sites would be ~20% microbial methane generated in the surface sediments, 65% microbial methane venting from accretionary prism sediments, and 15% thermogenic gas migrating from depth. The isotopic composition of the thermogenic gas suggests origin at temperatures of 125°–135°C, or depths of 2–2.2 km (Rooney et al., 1995).
Gas Transport through the Gas Hydrate Stability Zone
It is frequently proposed that a critically pressured free gas column can build up beneath a seal created by the base of gas hydrate stability (de Boer et al., 1985; Grauls, 2001; Hornbach et al., 2004; Flemings et al., 2003). This situation, however, would seem to be inconsistent with the concept of the base of gas hydrate stability being a pressure-temperature (P-T) equilibrium surface. If gas pressure builds up and excess water is present, then gas should react with water to form additional gas hydrate until the pressure is brought back to equilibrium for the prevailing temperature. The presence of free gas at high concentration in a confined migration pathway may reconcile this apparent contradiction.
At several Leg 204 sites where the ash/turbidite layer (Horizon A) was drilled and logged beneath the GHSZ, there was evidence for gas saturation 40%–80% of porosity (Tréhu, Bohrmann, Rack, Torres, et al., 2003). As discussed earlier, the gas in Horizon A has a characteristic chemical and isotopic composition that enables delineation of migration pathways. It appears that free gas is migrating through Horizon A to the base of the GHSZ, then through vertical seismic features (gas chimneys) to the seafloor. Estimated P-T conditions at the base of the GHSZ beneath southern Hydrate Ridge are 9.6 MPa and 11.3°C. Under these conditions (methane compressibility factor [z] = 0.85), porous rocks containing 65 vol% CH4 and 35 vol% water would have a CH4-H2O system composition of 14.4 mol%, equivalent to the formula CH4·5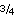 H2O. This suggests that when a gas stream migrates in the presence of <35% water saturation at these P-T conditions and moves into the GHSZ, available water converts to gas hydrate and a liquid phase, at least locally, would not be present. This creates conditions under which free gas and gas hydrate can coexist within the GHSZ. Moreover, it is an implication of the phase rule (number of components + 2 – number of phases = degrees of freedom) that the system gains an additional degree of freedom because of localized loss of the liquid phase so that gas pressure can rise above hydrostatic. In this view, system composition (high gas saturation at the base of the GHSZ) is the primary factor that allows both the existence of free gas and some degree of gas overpressure to occur at the base of and within the GHSZ.
H2O. This suggests that when a gas stream migrates in the presence of <35% water saturation at these P-T conditions and moves into the GHSZ, available water converts to gas hydrate and a liquid phase, at least locally, would not be present. This creates conditions under which free gas and gas hydrate can coexist within the GHSZ. Moreover, it is an implication of the phase rule (number of components + 2 – number of phases = degrees of freedom) that the system gains an additional degree of freedom because of localized loss of the liquid phase so that gas pressure can rise above hydrostatic. In this view, system composition (high gas saturation at the base of the GHSZ) is the primary factor that allows both the existence of free gas and some degree of gas overpressure to occur at the base of and within the GHSZ.
High salinities created when salt is excluded during gas hydrate formation (Torres et al., 2004) can also stabilize free gas at shallow depths within the GHSZ (Milkov et al., 2004b). There is no evidence, however, for high salinity at the base of gas hydrate stability or in the vertical gas chimneys as sampled at Site 1248.
Double Bottom-Simulating Reflector
Leg 204 seismic data show the presence of a faint, deeper, second BSR on the seaward side of southern Hydrate Ridge in places where Horizon A underlies the prominent regional BSR. This second BSR is imaged and noted in illustrations from the Leg 204 Initial Reports volume chapters for Site 1245 (fig. F3; Shipboard Scientific Party, 2003b), Site 1247 (fig. F3; Shipboard Scientific Party, 2003c), and Site 1248 (fig. F3; Shipboard Scientific Party, 2003d). The estimated depths, temperatures, and hydrostatic pressures at the upper (BSR1) and lower (BSR2) reflectors near the site locations are given in Table T8, along with the calculated gas hydrate decomposition temperatures for two different gas mixtures. The gas mixture used to calculate hydrate decomposition temperatures at BSR1 is a three-component mixture with a C1/C2 ratio of ~3000 and traces (a few parts per million) of propane (normalized molar fraction = C1:0.99961, C2:0.00033, and C3:0.00006). The gas mixture used to calculate BSR2 hydrate decomposition temperatures is a six-component mixture with a C1/C2 ratio of ~200 and significant amounts of C3–C5 hydrocarbons (normalized molar fraction = C1:0.9900, C2:0.005, C3:0.0025, i-C4:0.001, n-C4:0.001, and C5:0.0005). The first gas mixture is relatively pure methane, similar to that found in void gas samples in cores just above BSR1. The second is a wetter gas mixture characteristic of void gas in zones a few meters above and below Horizon A. Nonhydrocarbon gas components (O2, N2, and CO2) were neglected because they are believed to be mainly artifacts of air contamination and dissolved bicarbonate decomposition.
The calculations summarized in Table T8 show that the observed zonation of gas composition in Hydrate Ridge sediments is consistent with a GHSZ that is stable to greater depths and temperatures than relatively pure methane (Structure I) gas hydrate and with the occurrence of a second, deeper (Structure II) BSR. Obtaining these consistent results using the observed gas composition and the Colorado School of Mines Hydrate (CSMHYD; Sloan, 1998) computer program requires use of a uniform regional thermal gradient (58°C/km) that is the approximate mean of the corrected values measured at individual sites during Leg 204 (Tréhu, this volume).
Additional evidence for the existence of gas hydrate beneath the presumed Structure I gas hydrate BSR1 is threefold. First, some void gas samples collected from depths beneath BSR1 are highly enriched in propane, a possible consequence of voids that formed in the vicinity of decomposing Structure II gas hydrate. Examples are Sections 204-1245B-19X-3 and 204-1248C-15H-1. Structure II gas hydrates enclose gas that is significantly enriched in propane relative to the feed gas from which it was formed. These gas samples are present at depths near the computed depths of BSR2, where propane selectively removed from dissolved gas within the hydrate stability zone would have been released upon gas hydrate decomposition. Second, some headspace gas samples collected beneath BSR1 contained greater amounts of gas than would have been expected from residual dissolved gas. Examples are Sections 204-1245B-16X-4 and 204-1247B-22X-2. Third, some sediment samples collected beneath BSR1 for headspace gas analysis in a 1-cm-diameter brass cork-boring tube appeared to freeze inside the tube between the time of collection and displacement from the tube (with some difficulty) by means of a wooden dowel. Freezing of the water in the sediment could have been due to heat taken up by decomposition of Structure II gas hydrate.
Reaction-Dominated Gas Hydrates
In contrast to Leg 204 sites with evidence for migrated hydrocarbons, the gas geochemistry of cored sediments at Sites 1244, 1246, 1251, and 1252 and of shallow sediments at Sites 1245 and 1247 does not show obvious influence of migrated hydrocarbons. These sites are characterized by relatively steep concentration gradients in the microbial metabolites (SO42–, HCO3–, CH4, NH4+, and PO43–). Whereas diffusive flux obviously occurs, the gradients are primarily caused by rapid reactions driven by the decomposition of sedimentary organic matter. The gas hydrates that develop under these circumstances are referred to as reaction-dominated gas hydrates, in contrast with previously discussed transport-dominated gas hydrates.
Methane is generated in shallow anoxic sediments as part of the normal process of organic matter remineralization (Claypool and Kaplan, 1974). Microbial respiration processes are ubiquitous in sediments where an adequate supply of metabolizable organic matter is present. Respiration involves the transfer of electrons from chemically reduced substances to oxidized substances, with capture of some of the energy produced enabling continuation of life processes.
Microbial Redox Reactions
In the terminal stages of the complex process of organic matter remineralization in marine sediments, soluble intermediate compounds are fermented (anaerobically oxidized) according to the general reaction
-
2CH2O(NH3)0.15(H3PO4) 0.01 + 4H2O
2HCO3– + 0.3NH3 + 0.02H3PO4 + 8e– + 10H+,
shown here for organic matter with approximate Redfield stoichiometry. For each eight electrons produced, the main products of organic matter oxidation are 2 mol bicarbonate plus 0.3 mol ammonia and 20 mmol phosphate. This oxidation half-cell reaction only proceeds when coupled with electron removal (reduction) reactions. Aerobic respiration involves the reduction of oxygen:
-
2O2 + 8e– + 8H+ 4H2O.
In anoxic marine sediments, the main electron removal reactions are sulfate reduction,
-
SO42– + 8e– + 8H+ HS– + 3H2O + OH–,
and, after sulfate is exhausted, the reduction of carbon dioxide (or bicarbonate) to form methane,
-
HCO3– + 8e– + 8H+ CH4 + 2H2O + OH–.
In addition, an important reaction for carbon mass balance is the reaction of carbonate ion (or bicarbonate) with dissolved calcium and magnesium to form authigenic carbonate:
-
HCO3– + (Ca, Mg)2+ (Ca, Mg)CO3 + H+.
Under certain conditions (high flux of methane into sulfate reduction zone), it is also possible for methane to be anaerobically oxidized:
-
CH4 + 3H2O HCO3– + 8e– + 9H+,
when coupled with sulfate reduction. Note that anaerobic oxidation of methane produces only 1 mol bicarbonate (and no ammonia or phosphate) for each eight electrons produced.
The progress of these reactions can be followed as concentration and stable isotope changes with increasing depth downcore in marine sediments. The main complicating factors include simultaneously occurring reactions and diffusive transport of reactants and products. A major effect of these processes is the transfer of material from the solid sediment (organic matter and iron oxides) and overlying ocean (SO42–, Ca2+, and Mg2+) to the pore water and back to the solid sediment (as iron sulfides, authigenic carbonate, and methane hydrate). These transfers also involve some vertical redistribution of material as a result of the different burial rates of sediment and pore water induced by compaction.
Burial Velocities and Advection from Compaction
The water content of sediments generally decreases with increasing depth of burial because of compaction under the influence of gravity. In accretionary prisms, effects of horizontal forces can also enhance water loss. Compaction causes differences in the relative motion of the sediment grains and the entrained pore water, which has important consequences for early diagenetic reactions and formation of gas hydrates. Under normal circumstances, both sediment grains and pore water are being buried and moved downward relative to the upwardly moving seafloor or sediment/water interface. However, the sediment moves downward at a faster rate than the water, and viewed from the standpoint of a given sediment layer, pore water is being advected upward. One consequence of these differences in downward burial velocity is that pore water is older than the sediment layer containing it and has had a longer time for reactions to proceed than is indicated by the age of the sediment.
The methods to track the relative movements of sediments and pore water during compaction are outlined by Berner (1980). Although compaction and porosity reduction are continuous, the main loss of porosity occurs at shallow depths (uppermost 50–100 mbsf). Compared with porosity loss at shallow depth, compaction at depths greater than a few hundred meters can be considered negligible. Using these considerations, the burial velocities of sediment and pore water are
-
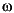 = [(1 –
= [(1 – 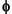 x)/(1 – )]x
x)/(1 – )]x
and
-
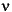 = (x/)x,
= (x/)x,
where
= burial velocity of sediment,
= burial velocity of pore water,
= porosity, and
x = value of porosity and burial velocity at depths where further compaction is negligible.
The burial velocities indicate time spent in a given depth interval, which can be integrated with depth to provide age vs. depth functions for the sediment and pore water. Figure F8 shows the porosity vs. depth and age vs. depth relationships for Site 1251. Porosity vs. depth is reasonably well described by = 0.79x–0.06, and this equation is used to calculate the burial velocities as a function of depth. A value of 500 m/m.y. as the sediment burial velocity (x) at depths where further porosity decrease is negligible enables matching of the sediment burial velocity with the independently determined biostratigraphic sedimentation rate (Tréhu, Bohrmann, Rack, Torres, et al., 2003). These differential burial velocities become important in calculating rates of diagenetic processes and understanding fractionation effects associated with transfer of material between the solid and aqueous or gaseous phases.
Sulfate Reduction and Evidence for Anaerobic Methane Oxidation
At sites near southern Hydrate Ridge but away from the summit, early diagenetic processes are best displayed by changes in pore water chemistry in sediments from the seafloor to ~50 mbsf. Sulfate concentration profiles for Sites 1244, 1245, 1246, 1247, 1251, and 1252 are shown in Figure F9. Sulfate gradients at summit Sites 1248, 1249, and 1250 could not be evaluated because sulfate was apparently depleted in the uppermost 15 cm of sediment beneath the seafloor (Boetius et al., 2000), a depth interval that is not well sampled in ODP operations. All of the sites away from the summit have similar shaped profiles, with sulfate concentrations greater than seawater frequently present in the upper 2–5 mbsf, then decreasing rapidly and approaching complete exhaustion at 5–12 mbsf. The dissolved sulfate profiles have an inverted S shape, being concave upward near the seafloor and concave downward at depths where sulfate approaches zero. The elevated sulfate concentrations near the seafloor suggest that some sulfide is being oxidized, possibly associated with bioirrigation or convective pumping of oxic bottom water to ~2–5 mbsf. The nonsummit site closest to the summit (Site 1247) has the deepest apparent influx of sulfate-containing water, whereas sites farthest from the summit (Sites 1251 and 1252) have the shallowest.
Sulfate profiles appear to be sublinear over the midrange concentration intervals (20–5 mM) but concave downward over lower ranges (5–0 mM). These dissolved sulfate gradients can be used to estimate net rates of sulfate reduction. Net rates calculated from concentration profiles are gross rates minus the rate of reoxidation (Jørgensen et al., 2004). Net rates are generally less than rates measured by radiotracer techniques. The rate of sulfate reduction can also be estimated from the total flux of sulfate from the overlying ocean into the sediment, or the diffusive flux plus the burial flux. This flux is equivalent to the depth-integrated net sulfate reduction rate (SRR) per unit area (Canfield, 1991):
-
Area SRR (mmol/m2/yr) = xDs(dC/dx) + oCo ,
where
x = average porosity,
o = initial porosity,
Ds = whole sediment diffusion coefficient for sulfate (average 6000 m2/m.y.),
dC/dx = sulfate concentration gradient (mmol/m4),
= estimated sedimentation rate (m/m.y.), and
Co = initial sulfate concentration (29,000 mmol/m3).
The apparent linear sulfate concentration gradients can be used directly to give estimates of integrated sulfate reduction rates. The sulfate concentration profile also can be fit with an exponential function and rates interpreted in the context of steady-state diagenesis (Berner, 1980):
-
C = (Co – C
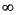 )exp[–(k/)x] + C ,
)exp[–(k/)x] + C ,
where
k = first-order rate constant,
x = depth in meters beneath the zone of surface sulfate penetration,
Co = initial sulfate concentration at the base of the bioirrigation or downwelling zone (29 mM) and,
C = projected asymptotic sulfate concentration where metabolizable organic matter would be exhausted.
In the case of Leg 204 nonsummit sites, Cx is a large negative number (e.g., –80 mM at Site 1244).
The rate of sulfate reduction within a given volume of sediment as a function of depth can be calculated from (Berner, 1980):
-
Volumetric SRR (mmol/m3/yr) = 0.5FkGoexp[(–k/)x],
where
0.5 = number of moles of sulfate reduced per mole of organic matter oxidized,
F = factor converting sediment from mass to volume concentration units (average = 1,400,000 g/m3), and
Go = concentration of metabolizable organic matter (mmol/g) calculated from (Berner, 1980),
-
Go = [(Co – C)(2 + kDs)]/20.5F.
Volumetric rates of sulfate reduction as a function of depth can be integrated over the depth interval where sulfate reduction takes place and compared with the integrated rate given by the linear curve fit. Table T9 summarizes estimates of net sulfate reduction rate parameters for Leg 204 sites away from the summit. Figure F10 shows the rate of sulfate reduction as a function of depth, along with the observed sulfate concentration profile and exponential fit to the data for Site 1244.
The integrated rates estimated from the exponential gradient are less than those estimated from the linear gradient, but both estimates are subject to uncertainties resulting from the number of factors and assumptions required in the calculations. All of the estimates are the same order of magnitude (14–50 mmol/m2/yr), with the differences being primarily because of differences in the biostratigraphic sedimentation rates used. The amounts of organic carbon needed to support the calculated rates of sulfate reduction are 0.3–0.5 wt%. Measured organic carbon contents (see individual site chapters in Tréhu, Bohrmann, Rack, Torres, et al., 2003) generally ranged from 0.9 to 1.6 wt%, with no obvious depth-related trends. Average values were 1.1–1.2 wt% at Sites 1245, 1246, 1247, and 1248 and 1.3–1.4 wt% at Sites 1244, 1250, and 1251. Organic carbon contents are mostly residual values measured at depths where intense organic matter remineralization processes are largely completed but in any case are adequate to support the observed extents of sulfate reduction and methanogenesis, as discussed below.
These rates of sulfate reduction can be compared with sulfate reduction rates in other environments. D'Hondt et al. (2004) found a net sulfate reduction rate of 25 mmol/m2/yr on the slope of the Peru margin, similar to what was observed at nonsummit sites near Hydrate Ridge. In contrast, Boetius et al. (2000) determined gross (radiotracer) volumetric sulfate reduction rates as a function of depth in the upper 15 cm of sediment at the summit of southern Hydrate Ridge. Observed rates beneath sulfide-oxidizing bacterial mats at the ridge crest were 0.5–5 µmol/cm3/d (1.4–14 mmol/cm3/yr), and the integrated rate over the 15-cm-thick sulfate reduction zone was 140 mmol/m2/d, (5.1 x 104 mmol/m2/yr), or more than 1000 times the calculated net rates of sulfate reduction at sites away from the summit of Hydrate Ridge. This rapid rate of sulfate reduction at the summit is due to the flux of gaseous methane to the seafloor, which supports sulfate reduction coupled with anaerobic methane oxidation (AMO) by the net reaction
-
CH4 + SO42– HCO3– + HS– + H2O.
Some supporting evidence for this process is the occurrence of isotopically light carbonate in the sediments (Greinert et al., 2001), which is a product of oxidized methane.
The pore water chemistry at Leg 204 nonsummit sites can be used to evaluate possible occurrence of methane oxidation. The 13C of DIC is shown in Figure F6. Only at Sites 1246 and 1245, with minimum 13C values of –30 and –24, respectively, is there possible isotopic evidence for some anaerobic methane oxidation. For example, at Site 1246 the minimum DIC 13C value of –30 could be produced by mixing ~12.5% DIC of –100 from oxidation of the shallowest, earliest-formed microbial methane (70 more negative than the DIC from which it is formed) with 87.5% of DIC of –20 from the oxidation of sedimentary organic matter.
The DIC 13C profiles in Figure F6 are typical of marine sediments in which anaerobic oxidation of organic matter is linked to bacterial sulfate reduction followed by carbonate reduction or methane generation. The DIC produced during sulfate reduction has the same 13C as the organic matter undergoing oxidation. Organic carbon in marine sediments of the northeast Pacific generally has 13C from –24 to –21 (Peters et al., 1978; Hedges et al., 1984; Dean et al., 1994). The DIC derived from oxidation of organic matter during sulfate reduction initially mixes with buried seawater DIC of ~0 and eventually reaches about the same 13C as the oxidized carbon source (Presley and Kaplan, 1968). At the depth where the dissolved sulfate supply is exhausted, the DIC 13C has a minimum 13C value then reverses and becomes heavier because carbon that is ~70 lighter than the DIC pool is being removed to make methane at a faster rate than DIC of –20 is being added (Claypool and Kaplan, 1974). If methane were being oxidized at the base of the sulfate reduction zone, the DIC 13C values would be much more negative (from –60 to –30) as observed in sediments at other locations where AMO is apparently taking place (Claypool and Threlkeld, 1983; Claypool et al., 1985, 2003; Vuletich et al., 1989; Blair and Aller, 1995; Burns, 1998; Borowski et al., 2000).
Linear sulfate gradients near the base of the sulfate reduction zone are commonly assumed to result from downward diffusive flux that is balanced by upward diffusive flux of methane, with anaerobic methane oxidation occurring at the base of the sulfate reduction zone (Borowski et al., 1996; Niewöhner et al., 1998; Dickens, 2001). However, equating methane flux with sulfate flux is only valid if there is no sulfate reduction fueled by oxidation of sedimentary organic matter (Haese et al., 2003). Sublinear sulfate gradients do not necessarily require or imply a corresponding equal upward flux of methane and consequent AMO.
In addition to the lack of isotopic evidence for AMO in the DIC 13C at most of the Leg 204 nonsummit sites, the pore water chemistry also indicates that oxidation of sedimentary organic matter is the main or exclusive electron donor for sulfate reduction. As indicated earlier, organic matter oxidation produces 2 mol bicarbonate and 0.2 mol ammonia in order to generate the eight electrons required to reduce 1 mol sulfate. Figure F11 shows that bicarbonate production (measured as alkalinity and corrected for cation depletion) is about twice the degree of sulfate depletion within the sulfate reduction zone at Sites 1244, 1245, and 1247. The fact that levels of ammonium and phosphate ions are directly proportional to degree of sulfate depletion also supports sedimentary organic matter oxidation, rather than methane oxidation, as the main source of the electrons used in sulfate reduction at the nonsummit sites.
Alkalinity Generation and Microbial Methanogenesis
Oxidation of organic matter in anoxic sediments is a continuous electron- and CO2-generating process as long as metabolizable organic matter, the required microbial community, and suitable oxidized substances (electron acceptors) are available. When dissolved sulfate is exhausted, dissolved CO2 becomes the next available electron acceptor (Claypool and Kaplan, 1974). The pH of pore water in marine sediments generally is buffered at 7.7–8.3 by reaction with carbonates and clays. In this pH range, carbonate speciation is such that ~98% of total dissolved CO2 is present as bicarbonate, and titration alkalinity is a useful proxy for bicarbonate or DIC.
At the depth where sulfate is exhausted, methane concentrations increase and DIC 13C shows a marked reversal (Fig. F6) consistent with the onset of methanogenesis and the removal of 12C-enriched CO2 to produce CH4. In addition, the alkalinity vs. depth plot generally shows a discontinuity or reversal in concentration at the depth where sulfate is exhausted because of CO2 removal by microbial methanogenesis. Measured alkalinity at any depth beneath the zone of sulfate reduction is the net of CO2 addition from organic matter oxidation minus CO2 removal as methane and authigenic carbonate. Alkalinity production resulting from organic matter oxidation can be projected from the modeled sulfate gradient. The difference between the initial seawater sulfate concentration and the asymptotic sulfate concentration that would have been attained when metabolizable organic matter was exhausted is one-half the projected asymptotic bicarbonate concentration, assuming a 2:1 bicarbonate to sulfate stoichiometry for organic matter oxidation. Therefore, the difference between the projected and observed alkalinity increase during organic matter oxidation is a measure of the amount of CO2 removed to form methane and authigenic carbonate. A minimum estimate of carbonate precipitation is provided by the cation deficit (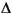 [Ca + Mg]) beneath the zone of sulfate reduction, and the amount of methane generated at a given depth can be estimated from the difference between total alkalinity generated minus the sum of remaining alkalinity plus the cation deficit, or
[Ca + Mg]) beneath the zone of sulfate reduction, and the amount of methane generated at a given depth can be estimated from the difference between total alkalinity generated minus the sum of remaining alkalinity plus the cation deficit, or
-
CH4 = Alktotal – (Alkmeasured + [Ca + Mg]).
Total alkalinity generated can be estimated from
-
Alktotal = (Cx – Co)(1 – exp[(–k/)x]) + Co,
where
Cx = asymptotic alkalinity (taken as twice the C value for sulfate reduction),
k = rate constant for organic matter oxidation (as determined from the sulfate gradient),
Co = initial bicarbonate concentration (2.4 mM),
= burial velocity, and
x = depth beneath the bioturbation or downwelling zone.
Figure F12 shows concentration trends with depth for dissolved methane and alkalinity at Site 1244. The smooth curve labeled Alktotal is an estimate of total alkalinity (bicarbonate) production as a function of depth, calculated from the equation given above. The irregular profile labeled Alkobsvd is the shipboard measured alkalinity, and the irregular profile labeled CH4(calculated) is methane concentration calculated from alkalinity mass balance. The curve labeled CH4(model) is from
-
CH4(model) = f(Alktotal)(1 – exp[–k/]x),
where
f = fraction of total alkalinity converted to methane (0.55–0.60) and
x = depth beneath the sulfate reduction zone.
Also shown on Figure F12 are methane phase relations (solubility curve from spreadsheet of Xu, 2002) and some direct measurements of methane content of Site 1244 sediments, which constrain the estimates of methane concentration presented here. Headspace gas analyses in the shallow sediments indicate that methane is absent within the sulfate reduction zone and begins to build up just beneath it. The six PCS cores show specific intervals of recovered methane content with depth in Site 1244 sediments. The two shallow PCS samples (24 and 40 mbsf) contain methane amounts consistent with only dissolved methane at concentration levels that are undersaturated with respect to methane hydrate (26 and 41 mmol/L of pore volume, respectively). Two intermediate-depth PCS samples (73 and 103 mbsf) have methane contents in excess of methane hydrate saturation (220 and 125 mM, respectively), which presumably consists of dissolved methane at saturation levels and small amounts (10 and 3 vol% of porosity, respectively) of methane hydrate. Three deeper PCS samples near the base of gas hydrate stability (120, 131 and 142 mbsf) are again undersaturated relative to methane hydrate and/or free gas. These PCS measurements are all consistent with the estimated quantities of methane generated as a function of depth in that PCS measurements indicate methane contents above saturation in the depth interval (44–113 mbsf) where the calculated methane contents exceed the theoretical solubility curve, and methane contents below saturation above and beneath this depth interval.
An additional constraint is provided by the observation that the shallowest chlorinity anomaly and occurrence of methane hydrate at Site 1244 was at ~45 mbsf (Milkov et al., 2004a; Tréhu et al., 2004). The calculated and modeled dissolved methane contents at Site 1244 also just exceeded saturation levels (~66 mM) at that same depth.
Similar results were obtained by modeling alkalinity and methane generation at the other nonsummit sites (1245, 1246, 1247, 1251, and 1252). The volumetric (mmol/m3/yr) rates of methane generation for Site 1244 are shown in Figure F13. These rates were integrated over interval 8–150 mbsf to give net integrated rates of methane production (mmol/m2/yr) for Leg 204 sites shown in Table T10.
Ethane Generation Mechanisms
Core void gas samples in marine sediments at shallow depths of burial are usually composed of methane (99.99+% on an air-free basis) with traces of ethane, propane, and carbon dioxide and variable air contamination. Ethane (C2H6) is the most abundant and frequently the only higher carbon–number (C2+) hydrocarbon detected in shipboard analyses of gases from cores. At least three mechanisms can account for ethane content of gases: microbial processes, low-temperature (<80°C) decomposition of organic matter, and migration of higher-temperature (>80°C) gas of thermogenic origin from deeper sediments. Evidence that microbial processes account for some ethane is the extremely light 13C-depleted ethane found in some gases of microbial origin (Mattavelli et al., 1992; Waseda and Didyk, 1995; Paull et al., 2000; Taylor et al., 2000). Microbial ethane could arise during methanogenesis if two-carbon organic acids (acetate and oxalate) can also be used, like CO2 as electron acceptors (Claypool, 1999). Evidence for microbial ethane from organic acid reduction is present in the form of isotopically heavy calcium oxalate minerals (Hoefs, 1969; Zak and Skala, 1993) and in organic acids in formation waters with carboxyl carbon having 13C as heavy as +38 (Franks et al., 1997). These could represent residual carbon pools that have undergone some conversion to ethane with a large kinetic isotopic fractionation. Alternatively, there is some evidence for enzyme-catalyzed carbon isotopic exchange between dissolved CO2 and carboxyl carbon of organic acids (O'Leary and Yapp, 1978). Such exchange would imply organic acids coexisting with dissolved CO2 depleted in 13C by methanogenesis, but not necessarily acting as alkane precursors.
Migrated ethane (usually accompanied by other C2+ alkanes) of obvious deep thermogenic origin is occasionally encountered at shallow depths in marine sediments at Hydrate Ridge and elsewhere (Schumacher and Abrams, 1996) and requires existence of some migration conduit.
The most common mechanism for ethane generation in marine sediments at relatively shallow (<1 km) depths of burial is low-temperature decomposition of sedimentary organic matter. DSDP/ODP cored sediments almost invariably show an exponential increase in ethane (as shown by C1/C2) with increasing temperature and depth of burial. The increase in C1/C2 ratio in core gas void samples from sediments that contain microbial methane is directly related to temperature history of the sediment. A simple kinetic calculation can reproduce most observed trends of C1/C2 in a range of sedimentary environments with widely varying temperature histories (Claypool, 1974). The inputs for such a calculation are sedimentation rate and geothermal gradient (heating rate) and kinetic constants for low-temperature ethane generation (A = 1014/s, E = 10 kJ/mol). Examples of observed C1/C2 ratios in sediments with differing thermal histories are illustrated in Figure F14. The appearance of increasing amounts of ethane and propane is frequently interpreted as a flux of thermogenic gas from deeply buried sediments; however, this interpretation is only warranted if quantities of ethane exceed that expected from the low-temperature decomposition of the buried organic matter.
Possible groupings of ethane of different origins according to 13C and C1/C2 are shown in Figure F4. Samples that contain large amounts of microbial ethane have 13C values from –55 to –45 and C1/C2 ratios >20,000. Microbial ethane is mostly in samples of hydrate-bound gases with no influence of migrated thermogenic ethane. The samples representing proposed low-temperature thermogenic or diagenetic ethane are mainly void gas samples from the slope basin sites (1244, 1251, and 1252). Samples dominated by migrated thermogenic ethane were from core gas voids at depths in and near Horizon A at Sites 1245, 1247, and 1248. The samples that plot between the diagenetic and thermogenic ethane fields are mostly near-surface hydrate-bound gas from the summit sites (1249 and 1250) and probably represent thermogenic ethane overprinted or mixed with some diagenetic or microbial ethane.
Ethane Fractionation by Gas Hydrate Formation
High-resolution gas geochemistry during Leg 204 showed that gases within the GHSZ at some nonsummit sites do not have the expected increase in ethane (i.e., are depleted in ethane), whereas those just below the GHSZ show a sharp (~10x) increase in ethane content (Tréhu, Bohrmann, Rack, Torres, et al., 2003; Milkov et al., 2004a). The ethane depletion of void gas samples within the GHSZ is associated with gas hydrate occurrence and is due to formation of Structure I gas hydrates, which are enriched in ethane relative to the dissolved gas from which they are formed (Sloan, 1998). Gas samples from core voids formed within the GHSZ were mainly from residual dissolved methane that is depleted in ethane resulting from gas hydrate formation, although some core voids at the top of the gas hydrate occurrence zone were from decomposed gas hydrate and showed some relative C2 enrichment (Milkov et al., 2004a). This molecular fractionation characteristic of gas hydrate occurrence is most apparent in the shallow microbial gas in slope basin sediments adjacent to Hydrate Ridge, where there is no influx of gas from the deeper subsurface (Milkov et al., 2005).
Such departure from the normal trend of absolute and relative ethane content is illustrated in Figure F15B and F15C for analyses of void gas samples from Sites 1244 and 1251. Here there are major discontinuities in ethane content occurring at depths corresponding to the pressure-temperature base of gas hydrate stability at each site.
Ethane enrichment in void gas samples below the base of gas hydrate stability was also observed during Leg 204 at Sites 1245, 1247, 1248, and 1250 (Tréhu, Bohrmann, Rack, Torres, et al., 2003). However, ethane distribution at these other Leg 204 sites is complicated by the presence of migrated thermogenic hydrocarbons (Milkov et al., 2005).
Comparison of C1/C2 in void gases and in hydrate-bound gases indicates that gas hydrate shows the expected C2 enrichment. Moreover, hydrate-bound gases do not contain C3, which is excluded by Structure I gas hydrate (Sloan, 1998). Calculations using the CSMHYD program (Sloan, 1998) show that the mole fraction of C2 in gas hydrate has a four-fold enrichment over the mole fraction of C2 in the gas from which the gas hydrate was formed.
The solid line in the C1/C2 plots of Figure F15A, F15B, and F15C shows the expected C1/C2 trends based on heating rate at Sites 1244, 1251, and 1252. The void gas samples from Site 1252 generally follow the expected trend with no obvious C2 depletions or enrichments above or below the base of gas hydrate stability. At Site 1252, there is no BSR, no chlorinity anomalies, and apparently little or no significant development of gas hydrates, although adequate CH4 contents were estimated by the model discussed above. In contrast at Sites 1244 and 1251 and compared with the expected trend of C1/C2, there is apparent C2 depletion above and enrichment below the base of gas hydrate stability, producing a significant offset in C1/C2 ratio at the respective depths (125 and 195 mbsf) of the base of gas hydrate stability. Void gas samples within the GHSZ are depleted in C2 because it has been selectively removed from the dissolved gas in the pore water and is being stored as gas hydrate in the solid phase of the sediment. As the sediment layers subside beneath the P-T horizon, marking the base of gas hydrate stability, the gas hydrate decomposes and releases C2-enriched gas, causing the observed relative C2 enrichment in the dissolved (or free) gas below the GHSZ at Sites 1251 and 1244.
The chemical fractionation at the base of gas hydrate stability is enhanced by the fact that a C2-enriched solid (gas hydrate) subsides more rapidly than pore water with dissolved gas because of compaction. This differential subsidence effectively transfers ethane from the dissolved phase above the base of gas hydrate stability to the dissolved (and gas) phase beneath the base of gas hydrate stability.
The discontinuity in C1/C2 at the base of gas hydrate stability could be developed as a quantitative indicator of gas hydrate abundance and is present in some previously cored ODP holes, such as at ODP Site 1019 in the Eel River Basin (Fig. F16). However, the C1/C2 discontinuity has not been observed at some other sites where gas hydrates were present, in part because of insufficient sample frequency, but probably also because of differences in the mode of gas hydrate formation.
