-
Q = –K x A x (
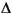 h/l),
h/l),
In the constant-pressure difference method, the hydraulic gradient across the sample is held constant while the flow rate is measured. The constant-flow approach measures hydraulic gradient while fluid is pumped into and out of the sample at a specified rate. The constant-flow approach is preferable at low flow rates because pressure differences can be measured with greater accuracy than flow rates. Based on previous permeability measurements (Saffer et al., 2000), permeabilities for the pelagic carbonate sediments were expected to be significantly higher than those of the hemipelagic sediments. Accordingly, constant-pressure difference tests were applied to the pelagic sediments, whereas constant-flow tests were conducted on the hemipelagic core samples.
The permeability tests were conducted using the Trautwein Soil Testing Equipment Company's DigiFlow K (Fig. F2). The equipment consists of a cell (to contain the sample and provide isostatic effective stress) and three pumps (sample top pump, sample bottom pump, and cell pump). Bladder accumulators allowed deionized water to be the fluid in the pumps while an idealized solution of seawater (25 g NaCl + 8 g MgSO4 per liter of water) permeated the sample. American Society for Testing and Materials (ASTM) designation D5084-90 (ASTM International, 1990) was used as a guideline for general procedures.
The Leg 170 and 205 core samples were stored in plastic core liners and sealed with wax during the cruise immediately after sampling to prevent moisture loss. The sealed samples were stored in the refrigerator at 4°C until immediately prior to sample preparation. Immediately before testing, cores were trimmed on both ends to provide freshly exposed surfaces using a wire saw or a utility knife, depending on core. After trimming the ends of the sample, the diameter and the height of the sample were measured (Table T1). The sample was then placed in a flexible wall membrane and fitted with filter paper and saturated porous disks on both ends. Next, the sample was placed in the cell, which was filled with deionized water so that the membrane-encased sample was surrounded by fluid. A small confining pressure of ~0.03 MPa (5 psi) was applied. Flow lines were flushed to remove any trapped air bubbles. After flushing the flow lines, the sample was backpressured at ~0.28 MPa (40 psi) in order to fully saturate the sample. Backpressure was achieved by concurrently ramping the cell pressure and the sample pressure to maintain a steady effective stress of 0.03 MPa. Saturation was verified by measuring the ratio of change in pore water pressure in the porous material to the change in the confining pressure (ASTM International, 1990). Once the sample reached saturation, the cell fluid pressure was increased while the sample backpressure was maintained, thus increasing the effective stress on the sample. Once the target effective stress was achieved, cell pressure and backpressure were maintained. The sample was allowed to equilibrate for at least 4 hr, generally overnight (12 hr).
After the target effective stress level was achieved, constant-pressure gradient tests were conducted (for the pelagic sediments) or one constant-pressure test was conducted to select an appropriate flow rate for the subsequent constant-flow tests (for the hemipelagic sediments). During the constant-flow tests, flow rates were maintained by the top and bottom pumps, one on each end of the sample, ensuring that the volume of the sample was unchanged. Since fluid pressure in the closed hydraulic system was affected by temperature changes, testing was conducted within a closed cabinet with a fan to keep the internal temperature uniform. The temperature was maintained at ~30°C (±1°C) during flow tests and consolidation steps, and temperature was monitored throughout the testing phase.
At least three permeability tests were performed at each effective stress level. Additional permeability tests were conducted on the pelagic carbonate samples because testing reached equilibrium flow rates very quickly. Once permeability values were obtained, cell pressure was increased and the sample was allowed to equilibrate overnight at the new effective stress. For every sample, three effective stress steps were performed, with values ranging from 0.14 to 0.62 MPa.
Using the flow rate (Q) and the pressure difference, hydraulic conductivity (K) values were calculated for each sample using Darcy's law:
h/l),
where
A = cross-sectional area of the sample,
h = difference in head across the sample (in meters), and
l = length of the sample.
Hydraulic head difference is related to pressure difference by:
h = z + P/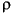 g,
g,
where
= density (1020 kg/m3),
z = elevation difference between the sample input and output.
Because the sample was vertical, z is equal to l, but with the sign depending on the flow direction. The density value was estimated for a temperature of 30°C and a salinity of 33 kg/m3, using the equation developed by Fofonoff (1985). Assuming a reasonable water compressibility, volume change, and therefore density change, due to the applied pressure is minor (<0.1%).
The hydraulic conductivity values were then converted to permeability (in square meters) using the following equation:
x g),
where µ = viscosity. A viscosity value of 0.0008 Pa·s was selected based on information from the Handbook of Chemistry and Physics (Lide, 2000) for water at a temperature of 30°C and salinity of 35 kg/m3. The average permeability was computed as the arithmetic mean of permeability values at each effective stress.
The corresponding porosity for each estimated permeability was calculated using the change in volume of fluid contained in the cell after each consolidation step. Using change in cell fluid volume accounts for both radius and length changes of the sample during consolidation. Total initial sample volume (VT(0)) was calculated using 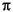 r2h, where r is the radius of the core sample and h is the height of the sample. Initial porosities (n0) for volume calculations were obtained from shipboard measurements (Kimura, Silver, Blum, et al., 1997; Morris, Villinger, Klaus, et al., 2003). Because a shipboard measurement could not be taken near Sample 205-1255A-4R-CC because of limited core, data from a similar depth at Site 1043 were used (Kimura, Silver, Blum, et al., 1997). Because the applied effective stress during backpressure is small (0.03 MPa), we assumed that the porosity of the sample at the end of backpressure is similar to the initial porosity (n0) of the sample.
r2h, where r is the radius of the core sample and h is the height of the sample. Initial porosities (n0) for volume calculations were obtained from shipboard measurements (Kimura, Silver, Blum, et al., 1997; Morris, Villinger, Klaus, et al., 2003). Because a shipboard measurement could not be taken near Sample 205-1255A-4R-CC because of limited core, data from a similar depth at Site 1043 were used (Kimura, Silver, Blum, et al., 1997). Because the applied effective stress during backpressure is small (0.03 MPa), we assumed that the porosity of the sample at the end of backpressure is similar to the initial porosity (n0) of the sample.
Using the initial porosity (n0), volume of voids before the testing (VV(0)) was calculated by multiplying the initial porosity by the sample volume and the volume of solids by subtracting volume of voids from sample volume. Using the difference of cell volumes between two consecutive steps (e.g., cell volume at backpressure and cell volume at first consolidation), the change in volume of water in the cell (VT(1)) and the new total volume of the sample was calculated. Using the calculated new total volume of the sample (VT(1)), the new porosity at the end of the consolidation is calculated. The new porosity (n1) at the end of the consolidation is:
A possible source of error in the permeability measurements is sample disturbance, in which drilling, recovery, and sample preparation may impact the flow pathways. Constant-pressure and constant-flow tests rely on the sample reaching equilibrium. Especially with lower permeability samples, error could be introduced by the sample not having fully equilibrated. Porosity is variable in the Costa Rica sediments (Kimura, Silver, Blum, et al., 1997; Morris, Villinger, Klaus, et al., 2003), and thus the shipboard porosity used may not be representative of the permeability sample. Although we attempted to subsample representative material for the grain size and carbon analyses, it is possible that variations in the core cause it not to be representative.
Approximately 15–20 g of sample was weighed into a beaker and treated with 3% H2O2 for a minimum of 24 hr until digestion of organic matter ceased. Next, the samples were dispersed with 250 mL of 4 g/L sodium hexametaphosphate (Calgon) for a minimum of 24 hr. All samples were further dispersed by 20–30 min of exposure in an ultrasonic bath, whereas the highly indurated samples were exposed longer with frequent stirring. The sand fraction was then separated from silt and clay fractions by wet sieving the sample through a 63-µm sieve. The silt and clay fraction was washed through the sieve into a 1-L cylinder. The sand fraction was rinsed with distilled water and transferred into a preweighed beaker. The sand fraction was then placed in a 70°C oven for drying. The dry sand fraction was weighed and the weight of the beaker was subtracted to calculate the total mass of the sand. To estimate the fine grain fraction (silt + clay), the fine grain suspension was brought to 1000 mL by adding distilled water and vigorously agitating. Using the settling velocities based on Stokes law, a 20-mL aliquot was extracted using a pipette from a depth of 20 cm after 53 s. The pipette was drained into a preweighed petri dish. The pipette was rinsed with distilled water and redrained into the petri dish. The petri dish was then placed in a 70°C oven for drying. The dried sample was weighed and the weight of the petri dish was subtracted to obtained the weight of the fine grain fraction. We corrected for the presence of Calgon in each aliquot by subtracting 0.02 g, and the result was multiplied by 50 in order to obtain a weight for the fine grain fraction without organic matter. To estimate the clay fraction, the remaining 980 mL of suspension was re-agitated and left to settle. After 1 hr 53 min 14 s, a 20-mL aliquot was extracted from a depth of 10 cm. As described above, the 20 mL sample was dried, weighed, and multiplied by 49 to obtain a weight for the clay fraction without organic matter. The silt fraction was estimated by subtracting the clay fraction from the fine grain fraction.
Approximately 15–17 mg of the dried and powdered samples was placed in polytetrafluoroethylene (PTFE) capsules for the total inorganic carbon (TIC; carbonate) analyses. TIC in the sediments was measured coulometrically (Engleman et al., 1985) with a coulometer coupled with an automated TIC preparation device that used 2-N perchloric acid to evolve CO2. Total weight percent carbon (TC) was measured using a Carlo Erba NA1500 carbon-nitrogen-sulfur elemental analyzer. The dried and powdered sample, in the range of 3–5 mg, was weighed into tin capsules, which are crushed and placed in a 50-position autosampler carousel. After flash combustion in a quartz column containing chromium oxide and silvered cobaltous/cobaltic oxide at 1040°C in an oxygen-rich atmosphere, the sample gas is transported in a He carrier stream and passes through a hot reduction column (650°C) consisting of elemental copper to remove oxygen. The effluent stream then passes through a chemical (magnesium perchlorate) trap to remove water. The stream then passes through a 1.5-m gas chromatographic column at 55°C, which separates the N2 and CO2 gases. Finally, the gases pass through a thermal conductivity detector.
Total weight percent organic carbon was estimated by subtracting TIC from TC.
Total weight percent calcium carbonate (CaCO3) was calculated by multiplying TIC by 8.333.
![]()