INORGANIC GEOCHEMISTRY
Typically, shipboard interstitial water analyses were performed on 5- or 10-cm-long whole-round sections that were cut immediately after the core arrived on deck. Details of the sampling resolution are described in the individual site chapters of this volume. After extrusion from the core liner, the surface of each whole-round section was scraped with a spatula. In samples recovered by XCB and RCB drilling technology, care was taken to separate the so-called "biscuited" core samples from their associated slurry before squeezing. These steps are required to avoid contamination of the interstitial water signal by seawater.
Interstitial waters were collected using titanium squeezers that were modified after the standard stainless steel squeezer (Manheim and Sayles, 1974). Pressure up to 275 MPa (40,000 psi) was applied using a hydraulic press. Where necessary, the sample was left in the squeezer for an extended period (up to 24 hr) and the applied pressure was increased very slowly. This modified technique helped to maintain interstitial water yields at levels sufficient for the shipboard and shore-based analytical programs.
Interstitial waters were passed through prewashed Whatman number 1 filters fitted above a titanium screen and subsequently extruded into a plastic syringe attached to the bottom of the squeezer assembly. All interstitial water samples were double-filtered through 0.45-µm polycarbonate filters. Samples for shipboard analysis were stored in plastic vials pending analysis. Aliquots for future shore-based analyses were placed in glass ampules or plastic tubes and heat sealed or kept in precleaned polyethylene bottles and stored in a refrigerator. In cases where trace amounts of hydrogen sulfide were present, sulfide was trapped by the addition of 0.2 mL of a 1% zinc acetate solution.
Interstitial water samples were routinely analyzed for salinity as total dissolved solids with a Goldberg optical handheld refractometer. The pH and alkalinity were determined by Gran titration with a Brinkmann Instruments pH electrode and a Metrohm autotitrator. Dissolved chloride was determined by titration with silver nitrate. Ammonium and a few silica concentrations were determined by spectrophotometric methods (Gieskes et al., 1991) using a Milton Roy Spectronic 301 spectrophotometer equipped with a sample introduction system. Alkali (Li, Na, and K) and alkaline earth (Mg, Ca, Sr, and Ba) elements together with manganese, iron, and boron concentrations were determined by inductively coupled plasma–atomic emission spectroscopy (ICP-AES) following the general procedure outlined by Murray et al. (2000). Sulfate was analyzed as total dissolved sulfur and silica as total dissolved silica by ICP-AES. Details of the procedure are given below. In preparation for analysis by ICP-AES, aliquots of interstitial water were acidified with nitric acid and diluted tenfold with deionized water (0.5 mL of sample + 4.5 mL of deionized water). Analytical blanks were prepared identically by analyzing deionized water, which was acidified to matrix match the samples. At all sites, sodium was determined by charge balance calculation where
-
[Na+] = [Alk] – [K+] – 2 [Mg2+] – 2 [Ca2+] + [Cl–] + 2 [SO42–]
(Broecker and Peng, 1982). The chemical data for interstitial waters are reported in molar units. The reproducibility of results, determined via multiple determinations of the International Association for the Physical Sciences of the Ocean (IAPSO) standard seawater (alkalinity, Cl–, Ca2+, Mg2+, K+, and SO42–), spiked synthetic seawater (ICP-AES determinations), or through the use of a calibration curve (NH4+, HPO42–, and Si[OH]4), is available in Table T10.
Determination of Total Dissolved Sulfur and Total Dissolved Silica by ICP-AES
Sulfur and silica have strong emission lines that may be used for the determination of both elements in interstitial waters by ICP-AES. The method should allow the determination of trace quantities of sulfate (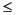 0.2 mM) and silica (100 µM) on relatively small sample volumes. For silica, two emission lines, at 212.4 and 251.6 nm, are recommended for analysis. We included both lines in the routine minor element run, which is performed on 1:10 diluted interstitial water samples. Calibration was performed by adding appropriate amounts of the silica standard to the master solution containing the other minor elements. The calibration curve covered a silica concentration range of 0–3340 µM. Twelve interstitial water samples were analyzed for silica by the routine shipboard colorimetric method. Figure F10A shows a comparison of results obtained by both methods, whereby the two emission lines mentioned for silica were used for analysis. Figure F10B shows that the results obtained for both silica emission lines are essentially identical. For this reason, the more sensitive silica emission line at 251.6 nm was used exclusively for further analysis.
0.2 mM) and silica (100 µM) on relatively small sample volumes. For silica, two emission lines, at 212.4 and 251.6 nm, are recommended for analysis. We included both lines in the routine minor element run, which is performed on 1:10 diluted interstitial water samples. Calibration was performed by adding appropriate amounts of the silica standard to the master solution containing the other minor elements. The calibration curve covered a silica concentration range of 0–3340 µM. Twelve interstitial water samples were analyzed for silica by the routine shipboard colorimetric method. Figure F10A shows a comparison of results obtained by both methods, whereby the two emission lines mentioned for silica were used for analysis. Figure F10B shows that the results obtained for both silica emission lines are essentially identical. For this reason, the more sensitive silica emission line at 251.6 nm was used exclusively for further analysis.
For sulfur, two strong emission lines at 180.676 and 181.978 nm (Jobin Yvon instrument library) were checked. According to other emission line libraries, the sulfur lines are located at 182.036 and 180.734 nm (B. Schnetger, pers. comm., 2003), but the latter line is interfered by calcium and manganese. Calibration curves were made up from IAPSO standard seawater diluted with a 3.5% NaCl solution made from Puratronic metal-free NaCl. In the high concentration range, both lines are suitable for analysis, but in the low concentration range (<5 mM), significant differences are evident. Results obtained from the sulfur line at 180.676 nm are significantly higher than those from the other line, most likely because of calcium interference. For this reason, only the sulfur line at 181.978 nm was used for subsequent analysis. In the ultraviolet range of the instrument, adequate flushing with nitrogen is required. This should be carried out for at least 12 hr prior to analysis. If this is not done, detection limits and reproducibility may be adversely affected. The ICP-AES method is very rapid (up to 30 samples may be analyzed per hour); therefore, we typically included a calibration point as every third sample.
