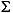 CO2 (TIC), assuming all TIC is present as pure CaCO3.
CO2 (TIC), assuming all TIC is present as pure CaCO3. In a very simplistic way, Demerara Rise sediments consist of variable mixtures of terrigenous detritus (represented by Al2O3 and SiO2) and biogenous material (represented by CaO and SiO2). To compare the relative proportions of the major components, the relative proportions of CaO (mostly carbonate), SiO2 (quartz/opal and alumosilicates), and Al2O3 (alumosilicates) are plotted in a triangle diagram (Fig. F2) (Brumsack, 1989). For comparison, average shale (AS) (Wedepohl, 1971), K-feldspar, and kaolinite are also plotted.
Sediments of lithologic Unit I (Fig. F2A) plot on a straight mixing line between carbonate and a clay component richer in Al than AS. This suggests that more intensely weathered clays (possibly a higher kaolinite proportion) are characteristic for Unit I sediments. Unit I samples show no additional enrichment in SiO2, ruling out that significant amounts of biogenic silica are present. Given the present lack of quantitative mineral data, the presence of biogenic silica could be obscured if a higher proportion of kaolinite were present.
Most sediments of Units II–IV (Fig. F2B) plot on a mixing line of AS and carbonate but show varying contents of biogenic silica. Unit II sediments are particularly rich in carbonate, whereas Unit III samples are generally lower in carbonate. The shift toward the SiO2 edge indicates higher excess silica contents.
Samples of Unit V (Fig. F2C) show a rather variable distribution. Excess silica contents are high in samples with low carbonate contents. This excess silica reflects the abundance of coarse-grained, quartz-rich sand in Unit V rather than to biogenous silica that typifies Units II–IV.
In Figure F3, we present an overview of the changing proportion of major components of all lithologic units. For calculations of major components listed in Table T1, we consider that
CO2 (TIC), assuming all TIC is present as pure CaCO3. Sediments from Demerara Rise are dominated by either carbonate or terrigenous detritus (Fig. F3). For lithologic Unit I, the terrigenous detritus forms the major component (>58 wt%). The average carbonate content is 37.0 wt%. The SiO2xs content (4.0 wt%) is the lowest found in any lithologic unit. Concentrations of OM and pyrite are <0.3 wt% and nonpyritic sulfur is absent in Unit I.
In Units II–III, carbonate (nannofossils, foraminifers, and partly diagenetic calcite) forms the major component, with concentrations that average 67.7 wt% in Unit II and 50.6 wt% in Unit III. SiO2xs contents (siliceous microfossils, radiolarians, and zeolites) are ~11 wt% in these sediments. According to low quantities of OM (<0.3 wt%), pyrite (<0.4 wt%), and nonpyritic sulfur (absent in Unit II, <0.1 wt% in Unit III), 20.1 wt% in Unit II and 38.1 wt% in Unit III of the sediment are terrigenous detritus.
The laminated black shales of Unit IV (of Cenomanian–Santonian age) are characterized by high OM contents (mean value >12 wt%, up to 24.9 wt% for individual samples). The content of pyrite is comparably low (0.7 wt%) in comparison to the amount of nonpyritic sulfur (2.1 wt%). This indicates a significant Fe limitation during pyrite formation and the sulfidation of OM. Beside the dilution effect of these components, the relative proportions of carbonate (48.4 wt%), terrigenous detritus (29.2 wt%), and SiO2xs (7.3 wt%) are similar to those found in overlying sediments of Unit III.
Like Unit I, Unit V is dominated by terrigenous detritus (57 wt%). The average carbonate concentrations (15.9 wt%) are the lowest found in lithologic units of Demerara Rise. In sediments of Unit V, siliceous microfossils are rare. Their former presence is indicated by zeolites. However, high amounts of SiO2xs (21.9 wt%) are mostly due to quartz, which is in accordance with the presumed shallow synrift deposit (Erbacher, Mosher, Malone, et al., 2004) (see discussion below). OM and pyrite contents are each 1.6 wt%. Thus, the pyrite content is highest in Unit V, whereas nonpyritic sulfur is present in smaller quantities (1.3 wt%) than in Unit IV.
The chemical index of alteration (CIA) (Taylor and McLennan, 1985) is a well-established parameter for determining the degree of weathering. During the degradation of feldspars, Ca, Na, and K are removed and clay minerals with a higher fraction of Al are formed. The CIA is estimated from the proportion of Al2O3 vs. the weathering-prone oxides:
where CaO* represents the amount of CaO incorporated in the silicate fraction. A correction for carbonate and apatite content is therefore necessary. Unaltered feldspars have a CIA of 50, whereas kaolinite has a value of 100 (total removal of alkali elements). We understand that CIA values in carbonate-rich sediments may lead to compromised results. The correction required for carbonate often leads to negative CaO* values because of the presence of additional carbonate phases like dolomite. In this case, CaO* contents were assumed to be zero. This may lead to an overestimation of CIA values due to an underestimation of CaO*.
As described above and shown in Figure F3 and Table T1, we assumed that the terrigenous detritus round off the major components we calculated from the chemical analyses to 100 wt%. When dividing the terrigenous detritus component of each sample by the Al2O3 content, a factor f is obtained, which represents the relative abundance of Al2O3 in this component. The reciprocal value of this factor 1/f (= weathering factor [WF]) should be a parameter for the degree of weathering, comparable to the CIA, but based on a broader range of chemical compounds.
In Figure F5 we compare WF with CIA values calculated for the lithologic units. Standard deviations (1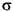 ) and results for AS (Wedepohl, 1971) are shown as well.
) and results for AS (Wedepohl, 1971) are shown as well.
Both parameters require a number of simplifications and assumptions and therefore bear uncertainties. The values of the CIA as well as WF spread within the different lithologic units. The standard deviations (<13 rel%) overlap for both parameters. Only qualitative interpretations of the mean values for each unit are possible. Nevertheless, the weak correlation between both parameters (R2 = 0.55) indicates that differences in weathering intensity did occur, since we can easily distinguish Units II–IV from Units I and V regarding their state of weathering.
For Units II, III, and IV, values for both parameters, CIA as well as WF, are essentially the same as for AS (CIA = 70–75) (Taylor and McLennan, 1985) and thus support our interpretation of Figure F2, where a terrigenous component similar in composition to AS is assumed.
The terrigenous material from Unit I is characterized by a CIA > 80, indicating a high degree of weathering. Taylor and McLennan (1985) report CIA values between 80 and 90 for the Amazon mud cone. This is confirmed by the WF. Deposition of lithologic Unit I began in the middle Miocene (Erbacher, Mosher, Malone, et al., 2004), which coincides with the initial uplift of the Andes. Resulting changes in provenance and/or drainage pathways (Potter, 1997) may have led to different characteristics of the terrigenous detritus in Unit I, in agreement with our observations from Figure F2.
In Unit V, where only one sample was encountered with CaO* contents >0 wt%, it is suggested that the CaO* content is underestimated and therefore the high CIA value is incorrect. WF indicates more intense weathering, but a clear conclusion cannot be drawn for such quartz-rich sediments.
In Figure F6, the concentrations of TiO2 (Fig. F6A) and Zr (Fig. F6B) are plotted vs. Al2O3. To avoid simple dilution effects when crosscorrelating element abundances, all samples were calculated on a "carbonate-free" basis. This calculation is based on the assumption that all TIC is present as pure CaCO3, which may lead to an underestimation for Ca and an overestimation for other carbonate-forming cations (mainly Mg and Sr, see discussion below). The resulting carbonate-free sediment still contains biogenous Si.
Data points from Units I–IV plot on a line (R2 = 0.93 for TiO2 and R2 = 0.65 for Zr), indicating that TiO2 and Zr are more or less uniformly incorporated into the clay component of these units. The observation that the AS data plot above the correlation line supports our interpretation that the terrigenous detrital component in Units I–IV is enriched in Al, possibly because of more intense weathering. Samples from Unit V plot above this line and show a negative correlation with Al2O3 (R2 = 0.74 for TiO2 and R2 = 0.86 for Zr). We conclude that an additional TiO2 - and Zr-bearing component other than clay minerals must be present in Unit V, most likely heavy minerals in the coarser-grained sands. In Figure F6C and F6D, the concentrations of TiO2 and Zr are plotted vs. SiO2xs (calculation as above but using carbonate-free data). Samples from Units I–IV again plot on a line (R2 = 0.53 for TiO2 and R2 = 0.33 for Zr). The negative correlation shows that SiO2xs behaves independently of the terrigenous-detrital component and is generally higher in samples with lower clay content. The same is essentially true for SiO2xs and Zr. By contrast, for samples from Unit V, a positive correlation is observed between SiO2xs and Zr (R2 = 0.86) or TiO2 (R2 = 0.82). We assume that the SiO2xs from Units I–IV is derived from biogenous Si, which serves as a diluent for the terrigenous component in the carbonate-free sediment, whereas TiO2 and Zr contents are higher in the quartz-rich sands of Unit V. Elevated quartz and heavy mineral abundances signify high-energy environments (Dellwig et al., 2000), supporting the idea that Unit V sediments are of synrift origin as stated by Erbacher, Mosher, Malone, et al. (2004).
The REEs are regarded as being almost insoluble and are present in only very low concentrations in seawater and river water (McLennan, 1989). Thus, the REEs present in sediments are mainly transported as particulate matter. Because the effects of diagenesis are minor, REEs reflect the chemistry of their source areas and can be used for provenance studies. Sedimentary sorting can affect the concentrations of REE: clays show higher abundances than do coarser-grained sediments. The relative composition of REEs are generally similar for sandstones and shales. Quartz has only a diluting effect. The presence of heavy minerals may have an effect on the REE composition of an individual sample; however, a large heavy mineral contribution would be required to significantly change distribution patterns. The REE compositions of biogenous carbonates and chemical sediments in general reflect the REE composition of the surrounding seawater (McLennan, 1989). But again, high quantities are necessary to cause changes in the REE character of the sediment relative to the primary detrital flux. The REEs have generally similar chemical and physical properties. This arises from the fact that they all form stable 3+ ions of similar size. A small number of the REEs also exist in oxidation states other than 3+, but the only ions of geological importance are Ce4+ and Eu2+. Changes in redox conditions can therefore affect the chemistry and thus the solubility of these two elements and lead to enrichments or deficiencies relative to other REEs. In a normalized REE distribution pattern, a positive or negative "anomaly" would result.
The average REE distribution patterns for the individual units are shown in Figure F7. To avoid dilution because of high carbonate contents, element/Al ratios are used. Elemental ratios are normalized to element/Al ratios of upper continental crust (Taylor and McLennan, 1985). Values between 1 and 2 for sediments of Units I–IV indicate a weak REE enrichment, whereas sediments of Unit V show a small depletion in REEs (values between 0.7 and 1). A slightly more pronounced negative Ce anomaly is seen in Unit IV relative to the other units. This Ce anomaly may be quantified by comparing the measured concentration (Ce) with an expected concentration (Ce*) obtained by interpolating between the values of the neighboring elements.
Wilde et al. (1996) linked Ce anomalies in shales of the anoxic facies to eustatic sea level changes. Similar to Mn, Ce4+ is less soluble under oxic conditions, whereas under anoxic conditions it will be mobilized, leading to a depletion in Ce in anoxic sediments relative to those deposited under oxic conditions. A negative Ce anomaly would result.
In Table T2, two different values are given for the Ce anomaly, which are based on different calculations. Taylor and McLennan (1985) recommended use of the geometric mean:
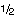 .
.
The ratio Ce/Ce* is then a measure of the anomaly, with values less than unity being termed negative. Wilde et al. (1996) support use of the arithmetic mean:
and calculated the logarithm of the ratio Ce/Ce*. Both calculations lead to essentially the same values for Ce*, with the most negative anomaly in Unit IV.
According to Wilde et al. (1996), the negative Ce anomaly for the black shales of Unit IV can be interpreted as a consequence of water column anoxia during sea level highstands in the Cretaceous. REE patterns in the sediments of Demerara Rise are complicated by the presence of biogenous and chemical compounds that record the surrounding seawater and pore water (e.g., for phosphate). For example, carbonate tests of plankton living in the photic zone under oxic conditions would carry the surface seawater characteristics and would therefore display a negative Ce anomaly. However, this effect is not seen in the more carbonate rich Units II and III.
The database is still too small for demonstrating that these REE characteristics hold true for Cenomanian/Turonian (C/T) black shales from Demerara Rise in general. Statistically, the REE patterns do not show any extraordinary characteristics and do not differ much from those of Post-Archean average Australian Shale (Taylor and McLennan, 1985).
Figure F8 illustrates the chemistry of the biogenous carbonates. In Figure F8A, CaO concentration is plotted vs. the TIC contents. The good correlation shows that almost all CaO is present as CaCO3. Some samples from Unit IV contain additional CaO, which is present as apatite. The contents of CaO-bearing mineral phases other than carbonate are negligible. The positive correlation between Sr and TIC (Fig. F8B) indicates that a variable fraction of Sr is incorporated into carbonates (750–1300 ppm). The negative correlation of MgO and TIC (Fig. F8C) shows that Mg is mostly incorporated into clay components, even though a small contribution of Mg-rich calcite or dolomite cannot be excluded. MnO shows no correlation with TIC (Fig. F8D), but highest concentrations of MnO are found in carbonate-rich Unit II. Figure F9 shows the average element/Al ratios of Ca, Sr, Mg, and Mn in Units I–V. Except for the strong Mn depletion in Unit IV (see discussion below), a similar distribution pattern is displayed.
The oxygenation state of the seawater and the redox state of the surface sediment form a crucial variable for the preservation of organic material and the formation of pyrite. Therefore, the contents of pyrite, reactive Fe, and TOC allow us to draw conclusions about the paleoenvironment during deposition of the sediment. The degree of pyritization based on bulk sediment analysis is visualized in a ternary Fex-TOC-S diagram (Fig. F10) (Brumsack et al., 1995). The content of reactive Fe (Fex) was estimated empirically (Fex = Fe – 0.25 x Al) assuming that a certain fraction of alumosilicate-bound Fe is not available for pyrite formation (Canfield et al., 1992). Results of a detailed analysis of reactive iron in the investigated samples is discussed in an accompanying publication (Böttcher et al., this volume). Data points that plot close to the pyrite saturation line (PSL) are assumed to represent samples that are completely pyritized. Most of the samples of Unit IV plot below the PSL, indicating the presence of an additional sulfur phase. Böttcher et al. (this volume) found organic sulfur contents exceeding 3 wt% in Demerara Rise black shales. The presence of acid volatile sulfur points to the presence of metal sulfides other than pyrite, likely ZnS (Brumsack, 1980). Samples of Units I–III are positioned above the PSL, indicating that Fex was only partly used for pyrite formation, in agreement with direct measurements of sulfur and iron speciation (Böttcher et al., this volume).
Trace metal (TM) distribution patterns reveal information about the depositional environment. Because of TM participation in biocycling processes (Bruland, 1983), scavenging by particulate matter and dissolution and precipitation of redox-sensitive compounds TM enrichment as well as depletion in sediments are diagnostic for bioproductivity and redox conditions during deposition. In combination with pore water data, they allow indication of postdepositional element migration. Figure F11 shows the mean values of TM/Al ratios of diagnostic TM in the different lithologic units. TMs are shown in order of TM enrichment relative to AS (dashed line) in Unit IV.
The nonlithologenic excess Ba has been interpreted as a paleoproxy for bioproductivity (Schmitz, 1987; Dymond et al., 1992; Paytan et al., 1996). These biogenic barites (BaSO4) (Bishop, 1988; Bertram and Cowen, 1997; Bernstein and Byrne, 2004) are only stable under seawater sulfate concentrations (Church and Wolgemuth, 1972). Because of the microbial sulfate reduction in TOC-rich sediments, barite is dissolved, Ba is mobilized (Brumsack and Gieskes, 1983; McManus et al., 1998; Eagle et al., 2003), and authigenic barite precipitates at the top of the sulfate-depletion zone forming diagenetic barite fronts within or above TOC-rich strata (Torres et al., 1996; Bréhéret and Brumsack, 2000).
Ba/Al ratios are highest in Units II and III. In the black shales of Unit IV, Ba/Al ratios are still high despite the absence of sulfate in the pore water (Erbacher, Mosher, Malone, et al., 2004). Arndt et al. (2006) show in a transport-reaction model that not only OM degradation but also anaerobic oxidation of methane above the black shales of Demerara Rise influence sulfate availability and therefore the remobilization of biogenic barium. The authors further showed that temporal dynamics of degradation processes caused various shifts of the barite precipitation zone during burial, thus inhibiting the formation of an authigenic barite front or causing the dissolution of earlier formed fronts. In our view, the Ba enrichment in Unit IV indicates elevated primary productivity during deposition. But a large fraction of former barite may remobilize and form diagenetic barites in Unit III. For this reason, the use of Ba as a paleoproxy on a quantitative level (Dymond et al., 1992) for Cretaceous settings in such an environment is highly questionable.
The very high phosphate contents (>0.7 wt% P2O5 on average in Unit IV) are comparable to those in recent upwelling sediments and also points toward enhanced nutrient supply and resulting high bioproductivity (e.g., Böning et al., 2004).
The black shales of Unit IV are clearly enriched in redox-sensitive and stable sulfide–forming TMs. In today's ocean, TOC-rich sediments are deposited in coastal upwelling areas and euxinic basins. Brumsack (2006) attempted to distinguish both environments by their specific TM patterns. Thereby, the author discussed TM sources and fixation mechanisms. Cu and Ni are discussed to be involved in biocycling. The enrichment found in recent upwelling sediments (Böning et al., 2004) indicates deposition via biodetritus. In contrast, oxyanions (As, U, V, and Mo) are primarily derived from seawater. The enrichments of Mo, U, and As indicate a sulfidic environment (Brumsack, 2006). High concentrations of sulfide-forming TM (Cu and Zn) and sulfur phases other than pyrite indicate Fe limitation and thus support the idea of an euxinic environment (Böttcher et al., this volume).
The strong depletion in Mn (Fig. F9) requires the presence of at least suboxic conditions in parts of the water column (Quinby-Hunt and Wilde, 1994). Dissolved Mn is conveyed away in an expanded oxygen minimum zone (OMZ) like in recent coastal upwelling areas. Thurow et al. (1992) describe such an Mn mobilization at the northwest Australian margin during the C/T boundary event. The authors found Mn-poor sediments within the OMZ and Mn-rich sediments below the OMZ, indicating oxic deep waters during the C/T interval at this location. Mn enrichment under anoxic/euxinic conditions is only possible in a closed (silled basin like) system, where dissolved Mn cannot be conveyed away and alkalinity is high enough to form Mn(II) carbonate. T.W. Lyons (pers. comm., 2006) finds these Mn-enrichments in euxinic Unit 1 in the modern Black Sea. Mn depletion in all samples of Unit IV shows that the investigated sites (Sites 1257–1261) must be located within the OMZ during deposition. In this case, the reoxidation and reduction of Mn oxides (Mn cycling) at the redox boundary may have induced TM scavenging by Mn (oxy)hydroxides (Cu, Mo, and V) comparable to the Black Sea. But the Mn most likely was deposited in other parts of the proto-Atlantic, where deep waters still might have contained oxygen.
However, one should mention that the elemental pattern of black shales from Demerara Rise is very similar to the one known from other C/T settings, particularly with respect to the extraordinarily high V and Zn contents (Brumsack, 2006).
![]()