INTRODUCTION
Stable isotope geochemistry of light elements (C, N, S, O, and H) is a recognized tool to reconstruct large-scale changes in global biogeochemical cycles from the Archaean onward. Over the past 5 yr, the stable isotopes of the transition elements have emerged as a novel and exciting development in the field of isotope geochemistry made possible by recent advances in multicollector inductively coupled plasma–mass spectrometers (ICP-MS). This is a rapidly expanding field of research that is only just beginning to address the physical, chemical, and biological driving forces for isotopic fractionation in nature. Iron is perhaps the most important transition element in (bio)geochemical processes and cycles. It is heavily implicated in contemporary paleoceanographic and paleoclimatological issues, such as the Fe fertilization hypothesis for the control of atmospheric carbon dioxide concentrations (Charette and Buesseler, 2000; Watson et al., 2000; Bakker et al., 2005).
There are four stable isotopes of Fe: 54Fe (5.84%), 56Fe (91.76%), 57Fe (2.12%), and 58Fe (0.28%), which show a mass-dependent fractionation of ~2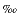 per atomic mass unit. Iron is a redox-sensitive element (Fe reduction is a common terminal electron accepting process), and so the changes in redox conditions that affect Fe speciation are of fundamental importance. However, there is only rudimentary knowledge about the mechanisms of kinetic isotopic fractionation in response to these changes. In addition, only sparse information exists on the equilibrium fractionation of Fe isotopes during crystallization of simple inorganic phases. As new experimental data become available, more Fe isotope measurements on samples from the natural environment are required to assess how closely natural systems can be modeled in terms of environmental changes, such as changes in Eh-pH and oceanic anoxia. In particular, natural environments for which a coherent set of proxy information already exists are the most suitable to examine the fractionation of Fe isotopes in nature.
per atomic mass unit. Iron is a redox-sensitive element (Fe reduction is a common terminal electron accepting process), and so the changes in redox conditions that affect Fe speciation are of fundamental importance. However, there is only rudimentary knowledge about the mechanisms of kinetic isotopic fractionation in response to these changes. In addition, only sparse information exists on the equilibrium fractionation of Fe isotopes during crystallization of simple inorganic phases. As new experimental data become available, more Fe isotope measurements on samples from the natural environment are required to assess how closely natural systems can be modeled in terms of environmental changes, such as changes in Eh-pH and oceanic anoxia. In particular, natural environments for which a coherent set of proxy information already exists are the most suitable to examine the fractionation of Fe isotopes in nature.
This preliminary study is concerned with the Fe isotope geochemistry in mid-Cretaceous organic-rich sediments at Demerara Rise and aims to explore whether Fe isotope geochemistry has potential to contribute to a better understanding of local and/or global redox processes during the mid-Cretaceous. Several ocean anoxic events (OAEs) occurred during the Jurassic and Cretaceous that represent periods with widespread anoxic conditions in the world's oceans and which had an impact on the oceanic Fe cycle. The Cenomanian–Turonian sequence on Demerara Rise recovered during Leg 207 was selected because it represents a combination of regional and global anoxia (Erbacher, Mosher, Malone, et al., 2004). Black shale deposition occurred throughout the Cenomanian and Turonian in a belt along the southern North Atlantic margin, while the world's oceans remained mainly oxic for most of this time interval. Global anoxia is represented by the latest Cenomanian OAE 2, when anoxic conditions were prevalent in much of the world's oceans. In theory, this makes the Demerara Rise sequence ideal to test if global vs. local effects on Fe isotope fractionation can be distinguished.
Our results are presented as a data report because only part of the planned measurements could be realized due to technical problems. These measurements, however, indicate that substantial fractionation of Fe isotopes did occur within the black shales, which in part can be correlated with a change in organic matter preservation. We investigated the possibility that these isotopic variations are influenced by changes in oceanic redox conditions.
Geochemistry of Fe Isotopes
Comparatively little is known about the possible fractionation mechanisms affecting Fe isotopes relative to our knowledge for isotopes of the lighter elements. With the exception of vital effects, temperature is the one principal control determining the magnitude of isotopic fractionation for C, N, O, and S. At atomic masses greater than ~50, the relative importance of temperature is diminished. Instead, other parameters known to play a subsidiary role for the lighter elements are implicated to have greater relative influence (Zhu et al., 2000). These include coordination chemistry, speciation, bond type, and redox conditions and can be generally categorized under "the bonding environment." Recent studies continue to reaffirm large, low-temperature Fe isotope variation. Implicit in these data is the importance of redox- and biologically induced fractionation in controlling the relative Fe isotopic abundance in natural reservoirs.
In many circumstances Fe(III) species are isotopically heavier than reduced Fe(II) species, although the reverse occurs during some reactions (Matthews et al., 2001). Under experimental conditions that simulate reaction pathways thought to play a role in natural environments, enrichments in 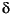 56Fe of 1.2–2.8 were found in Fe(III) relative to Fe(II) during inorganic and biological reactions (Beard et al., 1999; Anbar et al., 2000; Johnson et al., 2002; Croal et al., 2004; Icopini et al., 2004). Experiments have shown that for strictly inorganic systems, the bonding environment exerts a key influence in partitioning isotopes. But the relative dominance of kinetic or equilibrium processes remains poorly understood. Whereas it is relatively easy to generate a kinetic isotope fractionation during rapid precipitation of oxide minerals, experiments that demonstrate isotopic equilibrium are much harder to accomplish. So far, "equilibrium" fluid-mineral fractionations for Fe have been determined for
56Fe of 1.2–2.8 were found in Fe(III) relative to Fe(II) during inorganic and biological reactions (Beard et al., 1999; Anbar et al., 2000; Johnson et al., 2002; Croal et al., 2004; Icopini et al., 2004). Experiments have shown that for strictly inorganic systems, the bonding environment exerts a key influence in partitioning isotopes. But the relative dominance of kinetic or equilibrium processes remains poorly understood. Whereas it is relatively easy to generate a kinetic isotope fractionation during rapid precipitation of oxide minerals, experiments that demonstrate isotopic equilibrium are much harder to accomplish. So far, "equilibrium" fluid-mineral fractionations for Fe have been determined for
-
Fe(II)aq–hematite = –3.0 ± 0.3 (Skulan et al., 2002; Johnson
et al., 2002; Welch et al., 2003).
-
Fe(III)aq–hematite = –0.1 ± 0.2 (Skulan et al., 2002).
-
Fe(II)aq–siderite = 0.5 ± 0.2 (Wiesli et al., 2003).
-
Fe(II)aq–Fe(III) oxide = –0.9 ± 0.2 (Bullen et al., 2001).
-
Fe(III)aq–Fe(II)aq = 2.9 ± 0.2 (Johnson et al., 2002; Welch
et al., 2003).
-
Fe(II)aq–FeS = 0.3 ± 0.05 (Butler et al., 2003).
Theoretical predictions of isotopic fractionation based on 57Fe Mossbauer spectroscopic data (Polyakov and Mineev, 2000) have forecast 3–4 equilibrium isotopic fractionation (siderite-pyrite and siderite-magnetite). From the information above it is evident that relatively large Fe isotope fractionations take place during reactions that involve a change in redox state.
In general, only the reduced Fe species (Fe[II]) is soluble in the present-day oxic conditions of most surface environments, unless low pH conditions prevail. Despite this, the solubility of Fe(II) is very low in near-neutral pH environments, such that Fe concentrations <1 nM prevail in much of the modern open ocean (Martin and Gordon, 1988; Johnson et al., 1997) and exert one of the principal influences that limit marine productivity (Martin and Fitzwater, 1988). The residence time of Fe in the modern ocean is <200 yr, which is much shorter than the turnover rate of the global ocean (Broecker and Peng, 1982; Johnson et al., 1997). This implies that any fractionation of oceanic Fe will be dominated by local processes, as it does not remain long enough in solution for large-scale circulation processes to maintain isotopic contrasts between different water masses. Indeed, the Fe isotope composition of most modern Fe minerals is more or less constant at 0 and is indistinguishable from that of Fe in igneous rocks (Beard et al., 2003), indicating that isotope fractionation under modern (oxic) conditions plays a minor role. Loess, turbidites, fluvial particulates and organic carbon (Corg)-poor gray shales deposited under oxic conditions show a normal distribution of isotopic compositions with an average 56Fe of ~0, similar to terrestrial igneous rocks. The largest fractionation found in the modern ocean occurs in hot hydrothermal vents along mid-ocean ridges and in ferromanganese crusts (56Fe about –0.4 and about –0.6, respectively) (Beard et al., 2003; Levasseur et al., 2004).
In contrast, Corg-rich black shales deposited and diagenetically modified under anoxic conditions show a wide range in isotopic composition and include some of the most negative 56Fe values (Beard et al., 2003; Yamaguchi et al., 2003; Matthews et al., 2004). Fluctuations of 56Fe ~4 are measured in late Archaean rocks (–2.5–1.0 [Johnson et al., 2003] or –3.5 to +0.5 [Rouxel et al., 2005]), in which 56Fe values increase in the order pyrite-Fe carbonate-hematite-magnetite (Johnson et al., 2003). The large range of variability is attributed to a much larger pool of dissolved Fe(II), the result of the reducing conditions that existed in the late Archaean, whose isotope composition was fractionated globally by deposition of Fe oxides (Rouxel et al., 2005). The profound determining influence that redox has on the nature and abundance of Fe species and the large Fe isotopic fractionations that occur in response to changes in redox suggest that Fe isotopes may become a valuable proxy in paleoceanographic reconstructions.
