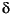 13C excursion defining OAE 2 is located between 424 and 426.5 mcd.
13C excursion defining OAE 2 is located between 424 and 426.5 mcd.
Samples were collected from the Cenomanian–Turonian black shale unit in Hole 1260B based on shipboard stratigraphic data (Erbacher, Mosher, Malone, et al., 2004). Ten samples were selected for Fe isotope analysis between ~404 and 456 meters composite depth (mcd), bracketing the late Cenomanian OAE 2. Age assignment of the samples (Fig. F1) is based on stable carbon isotope stratigraphy established subsequently (Erbacher et al., 2005), which showed that the 13C excursion defining OAE 2 is located between 424 and 426.5 mcd.
Bulk sediment samples were dried overnight at 50°C and powdered using an agate pestle and mortar. In these sediments, Fe is present within a number of mineralogical components, principally sulfide, carbonate, silicate, and oxide minerals. Three aliquots from the powdered samples were separated, and separate extractions were performed on each in order to extract Fe from the oxide-sulfide-carbonate and the oxide-only fractions, as well as a total digestion to extract Fe from all components except organic carbon (Tables T1, T2). The oxide fraction was selectively extracted using the Na citrate-Na bicarbonate-dithionite (CBD) (FeCBD) method (Hunt et al., 1995; van Oorschot and Dekkers, 1999), the oxide-sulfide-carbonate fraction was extracted using an aqua regia (AR) leach (FeAR), and the total digestion was performed using HNO3-HF (FeHF). All sediment residues were dried after extraction and weighed to determine the mass of sample dissolved.
The CBD method (Mehra and Jackson, 1960) is based on reductive Fe oxide dissolution with dithionite (Na2S2O4) as the reductant and Na citrate (Na3C6H5O7·2H2O) as a chelating agent to bind the dissolved Fe. Sodium bicarbonate (NaHCO3) is used to buffer the H+ loss during the reaction. The reductive solution of Fe oxides is a kinetic process and factors such as pH, crystallinity, and temperature have a major effect on the dissolution rate.
Samples dissolved in 7-M HCl were purified by anion exchange using analytical grade macroporous resin (AG MP-1), based on a procedure described by Maréchal et al. (1999) in which quantitative yields for Fe were reported. The majority of matrix elements were washed off the column with 24 mL of 7-M HCl, after which the Fe was eluted in 10 mL of 2-M HCl. The purified Fe solutions were evaporated to dryness, redissolved in 200 µL of concentrated HNO3, and made up to a volume of 10 mL with deionized water prior to screening for isobaric interferences on the masses of interest, particularly 54Cr, using a single-collector magnetic sector ICP-MS (Thermo Finnigan Element 2) operated in low- and medium-resolution modes. The raw data were blank corrected and normalized using indium (In) as the internal standard, from which Fe concentrations were calculated offline. Procedural blanks were always <36 ng Fe.
Iron isotope ratio measurements were performed using MC ICP-MS (Neptune, Thermo Finnigan) provided by High Lat resources (HPRI-CT-2001-00125) through the European Commissions "Access to Research Infrastructure" action of the Improving Potential Programme. The operating conditions adopted for the instrument follow the protocols described in Malinovsky et al. (2003) and Andrén et al. (2004). Detailed description of the procedure for optimization of ion lens settings and sampling depth in order to obtain lower instrumental uncertainty and higher mass bias stability is given in Andrén et al. (2004). The Neptune was operated in high-resolution mode (R = ~9000) throughout this study to resolve the Fe isotopes from the major spectral interferences:
The isotopes that were collected are 54(Fe + Cr), 56Fe, 57Fe, 58(Fe + Ni), 60Ni, and 62Ni. All analyses were made in an alternating sequence of samples and isotope standards (IRMM-014 Fe isotope standard). Maximum error in the 56Fe/54Fe ratio caused by 54Cr remaining after the anion exchange separation was assessed to be <0.005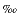 . No attempt was made to determine the 58Fe isotope because Ni was used as an internal standard for mass discrimination correction.
. No attempt was made to determine the 58Fe isotope because Ni was used as an internal standard for mass discrimination correction.
Samples and isotope standards were prepared at 5 ppm and spiked with 5 ppm Ni in 0.3-M HNO3. Ni doping allows a correction algorithm to be applied for mass discrimination effects in the plasma source using the mass discrimination factor (Ni) determined from the 62Ni/60Ni measurement. Linearity between the mass discrimination factors, Ni and Fe, is assumed constant during one measurement session according to the exponential correction employed (Woodhead, 2002), although between sessions the variation between mass discrimination factors can be significant. Data were corrected for mass discrimination effects using a procedure based on that described in Maréchal et al. (1999), Albarède et al. (2004), and Albarède and Beard (2004). By combining standard-sample bracketing with Ni doping (Anbar et al., 2001; Cardinal et al., 2003), mass discrimination factors are calculated for each standard-sample data set acquired over a short measurement interval, substantially reducing the effect on the data from variation in mass bias that occurs over time.
Isotopic data were processed using a combination of online baseline subtraction, calculation of ion beam intensity ratios, and outlier filtering by a 2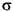 test. Further statistical treatment and mass bias corrections were performed offline. The internal analytical precision on the measured ratios was >50 ppm for the 56Fe/54Fe ratio and >100 ppm for the 57Fe/54Fe ratio at 1 standard deviation () level. The external precision on repeated measurements of the IRMM-014 Fe isotope standard using the raw 56Fe/54Fe ratios was typically ~450 ppm (2). Isotopic variation in 56Fe/54Fe is expressed as permil variation relative to IRMM-014 (56Fe/54Fe = 15.69859 ± 0.00027) using the standard notation (56Fe).
test. Further statistical treatment and mass bias corrections were performed offline. The internal analytical precision on the measured ratios was >50 ppm for the 56Fe/54Fe ratio and >100 ppm for the 57Fe/54Fe ratio at 1 standard deviation () level. The external precision on repeated measurements of the IRMM-014 Fe isotope standard using the raw 56Fe/54Fe ratios was typically ~450 ppm (2). Isotopic variation in 56Fe/54Fe is expressed as permil variation relative to IRMM-014 (56Fe/54Fe = 15.69859 ± 0.00027) using the standard notation (56Fe).
![]()