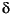 13C and 15N contained in the organic matter in the Blake Ridge sediments at Sites 994, 995, and 997 characterize the source of the sedimentary organic matter. Mean 13C values (-22.015N values (5.6
13C and 15N contained in the organic matter in the Blake Ridge sediments at Sites 994, 995, and 997 characterize the source of the sedimentary organic matter. Mean 13C values (-22.015N values (5.6
Measurements of the isotopic values of both 13C and 15N contained in the organic matter in the Blake Ridge sediments at Sites 994, 995, and 997 characterize the source of the sedimentary organic matter. Mean 13C values (-22.0![]() ±
1.4
±
1.4![]() ) and 15N values (5.6
) and 15N values (5.6![]() ±
0.7
±
0.7![]() ), plus the low C:N values (8 ± 2), indicate that the source of the sedimentary organic matter is largely marine (Meyers, 1994).
), plus the low C:N values (8 ± 2), indicate that the source of the sedimentary organic matter is largely marine (Meyers, 1994).
The stratigraphic uniformity of the organic carbon profile defines a constant baseline (i.e., -22.0![]() ±
1.4
±
1.4![]() ) for the organic carbon pool. The lack of significant stratigraphic variation in the 13C
(Fig. 2), 15N, and C:N values of bulk sedimentary organic carbon sampled from Blake Ridge sediments indicates that the character of the organic matter supplied to the Blake Ridge has not changed significantly throughout the sections sampled at Sites 994, 995, and 997. The variation in the isotopic composition of the host organic carbon is too small to be a major factor in generating the observed isotopic changes in the CH4 and dissolved CO2 pools.
) for the organic carbon pool. The lack of significant stratigraphic variation in the 13C
(Fig. 2), 15N, and C:N values of bulk sedimentary organic carbon sampled from Blake Ridge sediments indicates that the character of the organic matter supplied to the Blake Ridge has not changed significantly throughout the sections sampled at Sites 994, 995, and 997. The variation in the isotopic composition of the host organic carbon is too small to be a major factor in generating the observed isotopic changes in the CH4 and dissolved CO2 pools.
13C Values of CO2 Gas and DIC
A significant isotopic offset
(~12.5![]() ) exists between the 13C values of CO2 gas and DIC. A similar offset
(~10
) exists between the 13C values of CO2 gas and DIC. A similar offset
(~10![]() ) was reported between these pools at DSDP Site 533 (Claypool and Threlkeld, 1983). The offset is presumed to be an artifact of CO2 outgassing during sediment recovery. A significant equilibrium carbon isotope fractionation (8.38 ±
0.12
) was reported between these pools at DSDP Site 533 (Claypool and Threlkeld, 1983). The offset is presumed to be an artifact of CO2 outgassing during sediment recovery. A significant equilibrium carbon isotope fractionation (8.38 ±
0.12![]() , at 20° C; Emrich et al., 1970) occurs between bicarbonate and gaseous CO2. However, the observed 13C offset is larger than equilibrium predicts.
, at 20° C; Emrich et al., 1970) occurs between bicarbonate and gaseous CO2. However, the observed 13C offset is larger than equilibrium predicts.
Gas phases are only generated when saturation occurs during core recovery, largely as a consequence of the large decrease in pressure between the subseafloor and the surface. Because CH4 (Duan et al., 1992), but not CO2 (Weiss, 1974), becomes supersaturated as these cores are recovered, the majority of the original CH4 in the sediments below ~100 mbsf is lost during the core-recovery process (Dickens, et al., 1997). In fact, comparisons between the volume of gas in PCS samples and traditional headspace gas measurements below 300 mbsf indicate that ~99% of the original CH4 escapes during the recovery process. We infer that the DIC pool is sparged during CH4 degassing, and thus much of the original in situ DIC pool is lost as CO2 gas. A consequence of this loss is that the DIC and CO2 gas pools become fractionated relative to each other. Thus, the measured pore-water DIC isotope values reflect the residual DIC in the pore water after vigorous degassing and fractionation and not the isotopic composition of the original DIC pool.
The 13CDIC
values should accurately reflect the composition of the in situ DIC pool until CH4 saturation occurs (~30 mbsf). Below this level the original (predegassing) 13C values of the DIC pool probably lie somewhere between the measured CO2 gas and DIC values, but closer to the CO2 gas values, because these samples may more accurately reflect the majority of the original gas volume. In this paper, DIC 13C values are used to describe the changes in the CO2 pool in the upper 50 mbsf (the approximated depth at which visually obvious degassing started), and 13C values of CO2 gas are used below this level.
The distinct minimum of DIC carbon isotopic composition at ~20 mbsf is believed to be associated with a zone of active anaerobic methane oxidation (AMO; Reeburgh, 1976, 1980). This minimum corresponds with the sulfate-methane interface (Borowski et al., 1997). Within this AMO zone, 13C-depleted carbon obtained from CH4 oxidation is added to the DIC pool. Consequently, 13C-depleted DIC has diffused both upward and downward, depressing the 13C values significantly below the organic carbon baseline
(-22![]() ). The processes that occur at the sulfate-methane interface are treated in greater detail elsewhere (Borowski et al., 1997, and
Chap. 9, this volume).
). The processes that occur at the sulfate-methane interface are treated in greater detail elsewhere (Borowski et al., 1997, and
Chap. 9, this volume).
The 13C value of the shallowest CO2 gas sample from Site 994 (34.9 mbsf,
-20.6![]() ) is slightly 13C enriched compared to the sedimentary organic carbon base line. From this level down to ~120 mbsf, the 13C values of CO2 gas shift toward increasing 13C enrichment. The observed maximum in 13C values of both CO2 gas and DIC between 120 and 150 mbsf
(Fig. 2) requires addition of carbon with an isotopic composition enriched in 13C, and/or preferential subtraction of 13C-depleted carbon relative to the sedimentary organic carbon.
) is slightly 13C enriched compared to the sedimentary organic carbon base line. From this level down to ~120 mbsf, the 13C values of CO2 gas shift toward increasing 13C enrichment. The observed maximum in 13C values of both CO2 gas and DIC between 120 and 150 mbsf
(Fig. 2) requires addition of carbon with an isotopic composition enriched in 13C, and/or preferential subtraction of 13C-depleted carbon relative to the sedimentary organic carbon.
Although biogenic carbonates are enriched in 13C with respect to organic carbon (Rodriguez et al.,
Chap. 30, this volume), it is unlikely that the addition of carbon from the dissolution of biogenic carbonate is responsible for the 13C enrichment in the dissolved CO2 pool. The 13C value of the CO2 gas never gets more enriched than biogenic carbonate carbon
(~0![]() -2
-2![]() ), but does converge with the biogenic value. Thus, if biogenic carbonate was the source for the depleted carbon, the majority of the carbon in the CO2 gas would need to have come from biogenic carbonate dissolution. In contrast, the 13C values in the DIC samples are significantly enriched over the biogenic carbonate; thus their values cannot be explained by biogenic carbonate dissolution. Furthermore, carbonate textures and the pore-water chemistry suggest that carbonate precipitation, rather than dissolution, is occurring in this interval (Rodriguez et al.,
Chap. 30, this volume).
), but does converge with the biogenic value. Thus, if biogenic carbonate was the source for the depleted carbon, the majority of the carbon in the CO2 gas would need to have come from biogenic carbonate dissolution. In contrast, the 13C values in the DIC samples are significantly enriched over the biogenic carbonate; thus their values cannot be explained by biogenic carbonate dissolution. Furthermore, carbonate textures and the pore-water chemistry suggest that carbonate precipitation, rather than dissolution, is occurring in this interval (Rodriguez et al.,
Chap. 30, this volume).
The 13C enrichment in the CO2 pool probably results from fractionation of the dissolved CO2 pool during CH4 formation by CO2 reduction. The residual CO2 pool becomes 13C enriched as 12C-enriched DIC is selectively removed to form CH4 by CO2 reduction (Whiticar et al., 1986). Thus, the observed shift in the 13C values and resulting minimum at 120-150 mbsf is consistent with CH4 formation via CO2 reduction as previously inferred at Site 533 (Claypool and Threlkeld, 1983; Galimov and Kvenvolden, 1983).
We interpret the low molecular-weight hydrocarbon gases recovered during Leg 164 to be produced primarily by microbially mediated CO2 reduction. All the measured CH4 samples have
13CCH4
values more negative than -61![]() and previously reported methane to ethane ratios show a mean value of ~10,000 with a range of 700-39,000 (Paull, Matsumoto, Wallace, et al., 1996). These values are similar to those measured at Site 533 (Brooks et al., 1983) and consistent with CH4 of microbial origin
(Fig. 4). The range and mean values of both CH4 13C (-101.3
and previously reported methane to ethane ratios show a mean value of ~10,000 with a range of 700-39,000 (Paull, Matsumoto, Wallace, et al., 1996). These values are similar to those measured at Site 533 (Brooks et al., 1983) and consistent with CH4 of microbial origin
(Fig. 4). The range and mean values of both CH4 13C (-101.3![]() to
-61.1
to
-61.1![]() ; -68.4
; -68.4![]() ±
7.0
±
7.0![]() ) and D (-256
) and D (-256![]() to
-136
to
-136![]() ; -187
; -187![]() ±
20
±
20![]() ) (Fig.
5) are also consistent with the range of values characteristically associated with CH4 produced via CO2 reduction (Whiticar et al., 1986).
) (Fig.
5) are also consistent with the range of values characteristically associated with CH4 produced via CO2 reduction (Whiticar et al., 1986).
The causes of variation in ethane 13C values are not well established for biogenic gas deposits (Schoell, 1983), in part because ethane by definition comprises less than 0.1% of biogenic gas deposits (Bernard et al., 1977). However, small amounts of ethane are also produced by microbial processes (e.g., Vogel, 1982; Oremland et al., 1988). Whereas ethane 13C values typically are
5![]() -10
-10![]() enriched in 13C relative to methane, they ultimately are sensitive to the available organic carbon base line. The mean 13C value of the Blake Ridge ethane is ~44
enriched in 13C relative to methane, they ultimately are sensitive to the available organic carbon base line. The mean 13C value of the Blake Ridge ethane is ~44![]() depleted with respect to the host organic carbon. Similar fractionation elsewhere has been interpreted as being consistent with the ethane being of microbial origin (Waseda and Didyk, 1995).
depleted with respect to the host organic carbon. Similar fractionation elsewhere has been interpreted as being consistent with the ethane being of microbial origin (Waseda and Didyk, 1995).
The broad changes in the isotopic composition of CH4 reveal three subzones:
13CCH4
values are extremely depleted at the top of the methane-bearing zone (-10113CCH4
values are uniform (-64.0
Very negative 13CCH4
values are known to occur at the top of the methane-bearing zone throughout this region and numerous other similar locations worldwide (Borowski et al., 1997). The extremely depleted
13CCH4
values (e.g., ~-100![]() ) found near the sulfate-methane interface (SMI) are in part a response to local CH4 recycling (Borowski et al., 1997). At the SMI, extremely 13C-depleted CO2 (i.e.,
-37.7
) found near the sulfate-methane interface (SMI) are in part a response to local CH4 recycling (Borowski et al., 1997). At the SMI, extremely 13C-depleted CO2 (i.e.,
-37.7![]() , 20.40 mbsf, Site 995) is derived AMO. Thus, CH4 produced from this CO2 pool (via CO2 reduction) will exhibit progressively larger 13C depletion. Although, 13C depletion extends throughout the upper ~300 mbsf, it is implausible that adequate amounts of isotopically light, recycled CH4 produced via AMO are diffusing far enough downward, against the CH4 concentration gradient (Dickens et al., 1997), to produce the observed 13C trend between ~30 and 300 mbsf.
, 20.40 mbsf, Site 995) is derived AMO. Thus, CH4 produced from this CO2 pool (via CO2 reduction) will exhibit progressively larger 13C depletion. Although, 13C depletion extends throughout the upper ~300 mbsf, it is implausible that adequate amounts of isotopically light, recycled CH4 produced via AMO are diffusing far enough downward, against the CH4 concentration gradient (Dickens et al., 1997), to produce the observed 13C trend between ~30 and 300 mbsf.
The downhole shift in the CH4 isotope 13C values may reflect changes in the isotopic composition of the DIC pool as a consequence of CO2 reduction. A similar pattern seen at DSDP Site 533 was interpreted to be a result of Rayleigh fractionation associated with CO2 reduction (Claypool and Threlkeld, 1983).
Below ~300 mbsf, 13CCH4
values remain nearly constant (-64.0![]() ±
0.9
±
0.9![]() ) to the bottom of the holes (700-750 mbsf). Similarly,
13CCH4
values do not change throughout the holes. The uniformity in the isotopic composition of the CH4 pool below 300 mbsf suggests that the majority of the CH4 below this level was produced by the same process. No detectable change in the CH4 isotope values is associated with the BSR at ~450 mbsf (Holbrook et al., 1996).
) to the bottom of the holes (700-750 mbsf). Similarly,
13CCH4
values do not change throughout the holes. The uniformity in the isotopic composition of the CH4 pool below 300 mbsf suggests that the majority of the CH4 below this level was produced by the same process. No detectable change in the CH4 isotope values is associated with the BSR at ~450 mbsf (Holbrook et al., 1996).
Data from DSDP Site 533 suggests that the offset between
13CCH4
values and the 13C values of DIC and CO2 gas remain constant with depth (Galimov and Kvenvolden, 1983). However, the somewhat steeper geochemical gradients and the greater sample depths achieved during Leg 164 reveal a more complex depth relationship. Both the 13C values of CO2 gas and DIC from Sites 994, 995, and 997 clearly decrease with depth below 160 mbsf and thus the offset between the 13C values of CH4 and CO2 gas (or CH4 and DIC) also decrease with depth
(Fig. 6). The difference between the isotopic composition of the CO2 gas and CH4 is at a maximum of ~-72![]() at 120-150 mbsf
(Fig. 6). This difference is equivalent to a fractionation factor of 1.077, which is commonly observed for CH4 in marine sediments associated with CO2 reduction (Whiticar et al., 1986). This difference decreases with depth to ~-46
at 120-150 mbsf
(Fig. 6). This difference is equivalent to a fractionation factor of 1.077, which is commonly observed for CH4 in marine sediments associated with CO2 reduction (Whiticar et al., 1986). This difference decreases with depth to ~-46![]() at the base of the section, where the fractionation factor is 1.049, still within the range of fractionation factors that have been observed for CO2 reduction elsewhere (Whiticar et al., 1986; Whiticar, 1994). Therefore, this isotope data is consistent with CO2 reduction being the dominant methanogenic process in these sediments.
at the base of the section, where the fractionation factor is 1.049, still within the range of fractionation factors that have been observed for CO2 reduction elsewhere (Whiticar et al., 1986; Whiticar, 1994). Therefore, this isotope data is consistent with CO2 reduction being the dominant methanogenic process in these sediments.
Wellsbury et al. (1997) report that sediments at Site 995 contain surprisingly large amounts of acetate at depth. Acetate concentration is low near the surface (7 µM at 0.5 mbsf) and remains low for the next few hundred meters, but starts to increase sharply below 350 mbsf reaching 15,285 µM in the deepest sample (691 mbsf). Acetate is an important substrate for several microbial processes, hence, its interstitial concentrations are generally very low because of rapid turnover.
Wellsbury et al. (1997) interpreted the acetate inventories as being a consequence of increased bacterial activity at these depths, largely because of increased temperature. Laboratory experiments show that over short time periods, acetate formation is enhanced by sediment heating. Furthermore, they implicitly assume that the elevated acetate concentrations result in increased CH4 production.
One problem that Wellsbury et al. (1997) recognize with their interpretation is that their hypothesis predicts that a substantial contribution to the CH4 pool should come from acetate below 350 mbsf. Traditional interpretation of the expected range of CH4, D, and 13C values does not indicate CH4 production via acetate formation. In defense of this, Wellsbury et al. (1997) suggest there may be "a significant deuterium exchange during acetate methanogenesis" in situ, which does occur in culture experiments (e.g., de Graaf et al., 1996). Unfortunately, it is unclear the extent to which isotopic exchange observed during inherently rapid culture experiments reflects natural systems where the processes are much slower and the reactants are limited (e.g., Burke, 1993; Sugimoto and Wada, 1995). Although the robustness of using the D and 13C ranges to interpret the origins of the gas remains in question, we doubt that a significant shift from CH4 production by CO2 reduction to acetate fermentation would not result in a detectable affect in the CH4, D, and 13C values.
Although Wellsbury et al. (1997) were specifically discussing a different microbial population (e.g., acetate fermenters), the interpretation of increased microbial activity with depth conflicts with our data and model results. We suggest another explanation for the presence of acetate. Whereas the observed increase in acetate strongly suggests that some acetate production by microbial fermentation is continuing to occur within sediments at these burial depths, the increase in acetate concentration indicates that consumption of acetate is no longer occurring at rates sufficient to deplete the acetate pool size. Moreover, because the CH4 in the upper ~350 mbsf does not have an isotopic composition that reflects the acetate pathway, acetate production may be a relatively insignificant process throughout this sediment column. If so, the concentration of CH4 at depth is largely a consequence of CH4 migration, not local production (Paull et al., 1994).
Mass balance calculations indicate that the CH4 and CO2 reservoirs cannot be solely derived from the host sedimentary organic carbon. For example, if at 150 mbsf the CH4 carbon
(-71![]() ) and CO2 carbon
(-1
) and CO2 carbon
(-1![]() ) are derived from the sedimentary organic matter
(-22
) are derived from the sedimentary organic matter
(-22![]() ) in a simple closed system, then the concentration of CH4 should be ~30% of the total carbon gas pool. However, PCS data indicate that CH4 comprises ~95% of the total pool (Paull, Matsumoto, Wallace, et al., 1996). Thus, the composition of the CH4 and CO2 pools does not balance with the available organic carbon. Therefore, additional sources and/or sinks for carbon are important.
) in a simple closed system, then the concentration of CH4 should be ~30% of the total carbon gas pool. However, PCS data indicate that CH4 comprises ~95% of the total pool (Paull, Matsumoto, Wallace, et al., 1996). Thus, the composition of the CH4 and CO2 pools does not balance with the available organic carbon. Therefore, additional sources and/or sinks for carbon are important.
The only clearly identified sink in these sediments that is capable of removing a significant volume of isotopically enriched carbon is authigenic siderite formation (Rodriguez et al.,
Chap. 30, this volume). Siderite occurs in these holes below ~150 mbsf at the 2 to 4 wt% levels and has 13C values that range between 4![]() and
11
and
11![]() , with a mean value of ~+8
, with a mean value of ~+8![]() (Rodriguez et al., Chap. 30, this volume). Siderite formation does not result in significant carbon isotope fractionation. Thus, while removal of siderite carbon could produce a mass balance at ~150 mbsf, it does not help explain the smoothly varying carbon isotope profiles with depth.
(Rodriguez et al., Chap. 30, this volume). Siderite formation does not result in significant carbon isotope fractionation. Thus, while removal of siderite carbon could produce a mass balance at ~150 mbsf, it does not help explain the smoothly varying carbon isotope profiles with depth.
Conversely, CH4 and CO2 migration are potentially major sources of additional carbon. The PCS data show that the total methane and CO2 concentrations continue to increase below the base of gas hydrate stability (~450 mbsf) and remain high to the depth of the lowest successful PCS samples (560 mbsf) (Paull, Matsumoto, Wallace, et al., 1996; Dickens et al., 1997). Thus, CH4 and CO2 should at least be diffusing upwards along a concentration gradient from the underlying large gas reservoir.
A series of different isotope and mass balance models were run to simulate both closed- and open-system conditions with respect to observed 13C profiles for CH4 and CO2. All the models are based on initial concentrations and isotopic compositions of dissolved CH4 (15 mM and
-90![]() ) and CO2 (20 mM and
-22
) and CO2 (20 mM and
-22![]() ), like those measured at ~35 mbsf at Sites 994, 995, and 997. Changes in the isotopic composition of the carbon pools were computed using a Rayleigh fractionation model with a fractionation factor of 1.06, which is typical for CH4 production via CO2 reduction (Whiticar et al., 1986).
), like those measured at ~35 mbsf at Sites 994, 995, and 997. Changes in the isotopic composition of the carbon pools were computed using a Rayleigh fractionation model with a fractionation factor of 1.06, which is typical for CH4 production via CO2 reduction (Whiticar et al., 1986).
Our initial model further tested whether the concentrations and isotopic composition of the major carbon-bearing reservoirs (CH4 and CO2) could be derived in a closed system from CO2 reduction
(Fig. 7A). Closed-system model results failed to reproduce the observed isotopic values, concentrations, and profile curvatures. Moreover, addition of CO2 (to simulate the DIC increase that could occur within a closed system) through organic matter decomposition
(22![]() ) or carbonate dissolution
(~0
) or carbonate dissolution
(~0![]() ) does not improve model results. Previous attempts to model gas compositions from the Blake Ridge as a closed system at Site 533 also failed (Claypool and Threlkeld, 1983). Claypool and Threlkeld (1983) attributed the failure of the closed system model to the addition of gas from below.
) does not improve model results. Previous attempts to model gas compositions from the Blake Ridge as a closed system at Site 533 also failed (Claypool and Threlkeld, 1983). Claypool and Threlkeld (1983) attributed the failure of the closed system model to the addition of gas from below.
A second model considered the consequences of adding CH4 and CO2, thus simulating the effect of upward gas migration through the sediment column. In this migration model, a fixed amount of CH4 and CO2 were added at each step, and a constant proportion of the added CO2 was reduced to CH4 as if by CO2 reduction. Isotopic values were computed using a Rayleigh fractionation model (after Ussler and Paull, 1995), and isotope pool values were computed using a linear mass fraction mixing model (Gregory and Criss, 1986). This gas migration model
(Fig. 7B) replicates the main characteristics seen in the upper portion of the observed CH4-isotope profiles
(Fig. 2) when increasing amounts of carbon are added and when the majority (99%) of the CO2 is transferred to CH4 at each step. Lower transfer efficiencies cannot reproduce the curvature and light isotopic values either. Initial model values of the CO2 pool are also generally concordant with observed data. However, the consequence of the efficient CO2 to CH4 transfer is that the residual CO2 pool becomes unrealistically enriched in 13C as the model runs. Moreover, the 13C values of CO2 and CH4 values do not converge in this model, as they do in the observed data
(Fig. 2).
A third model simulates the effects of progressive reduction in microbial activity with depth. This model is the same as the second model except the transfer efficiency was incrementally decreased to zero (Fig. 7C). Model results reasonably reproduce the observed isotopic values, concentrations, and profile curvatures (Fig. 2). The results of these models indicate that the isotopic data can be best explained as a consequence of the incremental addition of both CO2 and CH4, along with a progressive decrease in microbial activity with depth.
![]()