- Rate = øDsdC/dx
+ øo
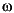 Co
- øbCb,
Co
- øbCb,
With the exception of Site 1002, the sites of Leg 165 were characterized by low TOC contents and, correspondingly, redox pathways dominated by suboxic diagenesis and low rates of bacterial sulfate (SO42-) reduction, an anaerobic process (Froelich et al., 1979). These reactions are recorded as enrichments in dissolved manganese and iron in the upper sediment layers and as decreases in dissolved sulfate downcore that vary in magnitude from site to site. Pore-water ammonium (NH4+) concentrations are strongly linked to release associated with decomposition of organic nitrogen compounds (Table 1). Nevertheless, mass balance considerations based on the reaction stoichiometry for bacterial sulfate reduction (assuming a Redfield C:N ratio), as well as the observed downcore variability in concentration, suggest that uptake of biologically liberated ammonium during silicate alteration (ion exchange reactions) is likely a factor (Berner, 1980). Downcore and intersite relationships for dissolved phosphate, although being sourced by organic decomposition, are complicated by the effective adsorption of phosphate onto the surfaces of carbonate sediment (Walter and Burton, 1986; Morse and Mackenzie, 1990).
Increases in pore-water manganese and iron record microbially mediated reductive dissolution of manganese and iron oxides and oxyhydroxides, whereas sinks are represented by incorporation into calcite (Dromgoole and Walter, 1990; Morse and Mackenzie, 1990) and by iron sulfide formation (Berner, 1984), albeit of generally minor extent for the latter. Downcore variability in the dissolved iron and manganese reflects nonsteady-state concentrations of TOC and the reactive solid phases of iron and manganese. These concentration relationships are linked in part to variations in calcium carbonate accumulation (i.e., varying dilution effects). Differences also reflect variation in the provenance of the solid manganese and iron as recorded in varying Mn/Ti and Fe/Ti ratios in the host sediments. Alkalinity (bicarbonate) levels are strongly influenced by production during organic decomposition (i.e., oxidation of organic matter), although carbonate reactions overprint the redox pathways (Table 1). Details of redox relationships are well represented in the data from Site 999 (Fig. 2) and in site chapters provided in Sigurdsson, Leckie, Acton, et al. (1997). Aspects of sulfate reduction will, however, be explored in further detail.
Generally low levels of sulfate reduction are corroborated by the paucity of methane in the headspace gases collected at the four sites (see exception at Site 1000 discussed below under the "Carbonate Reactions" section). The observed deficiencies in methane are expected given that appreciable methanogenesis occurs only after sulfate is depleted and sulfate reduction gives way to methane generation in the hierarchy of bacterial redox reactions observed in marine sediments (Martens and Berner, 1974; Froelich et al., 1979; Berner, 1980). To compare rates of sulfate reduction among Leg 165 sites and relative to deep-sea sediments throughout the world, depth-integrated rates (mmol SO42- cm-2 yr-1) were modeled at Sites 998, 999, 1000, 1001 (Berner, 1980; Canfield, 1991):
where ø is the average
porosity in the zone (depth interval) of sulfate reduction, and dC is taken as
the overall change in sulfate concentration over the zone of sulfate reduction (dx).
This approach simplifies the expression by assuming a linear decrease in sulfate
concentration over the interval of interest, which yields approximate and
minimum estimates that are sufficient for our general comparative purposes given
the shapes of the observed profiles. Co and Cb
are the concentrations of sulfate at the sediment/water interface (assumed to be
the seawater value of 28.9 mM) and at the base of the zone of sulfate reduction,
respectively. Porosity at the sediment/water interface and at the base of the
zone of sulfate reduction are represented by øo and øb,
respectively. Ds is the diffusion coefficient for dissolved sulfate
in sediment, which is affected by temperature and the tortuosity of the sediment
(Li and Gregory, 1974; Berner, 1980). Ultimately, the value for Ds
was carefully chosen (Table 2)
based on a recent evaluation of the effects of sediment type and porosity/tortuosity
(Iversen and Jørgensen, 1993). The assumption of a constant Ds value
within the zone of sulfate reduction is a simplification, given variations in
porosity as a function of depth, but is warranted in light of the objectives of
the study and the comparatively minor impact a more rigorous approach would have
on the conclusions. The average sedimentation rate over the zone of sulfate
reduction is indicated as ![]() .
.
These estimated parameters (based on data in Sigurdsson, Leckie, Acton, et al., 1997) and the calculated depth-integrated sulfate reduction rates are summarized in Table 2. The calculations are predicated on the assumption that infaunal mixing is not a factor over the zone of interest, which is justified given depths of tens to hundreds of meters for the zones of sulfate decrease at these sites (Canfield, 1991). The base of the zone of sulfate reduction is typically delineated by the attainment of asymptotic sulfate concentrations (Sites 999 and 1000); however, such concentrations are not observed over the analyzed intervals at Sites 998 and 1001. Consequently, the calculation can only be performed over zones of steadily decreasing sulfate for these latter sites and thus yields minimum values for the total depth-integrated rate of sulfate reduction. Furthermore, given the presence of a temporally significant unconformity at Site 1001, the sampled intervals above and below the hiatus were treated separately in the calculation and summed for a total rate (Table 2).
To test the validity of treating the sulfate trends (dC/dx) as linear, the sulfate profile from Site 1000 was subdivided into two distinct linear trends to account for the "steeper" sulfate depletion in the upper 50 meters below seafloor (mbsf) (Table 2). Of the four sites, this profile is least effectively represented by a single linear approximation, yet the calculated depth-integrated sulfate reduction rates differ only by a factor of 1.9 for this most extreme example: 9.3 x 10-5 mmol cm-2 yr-1 vs. 18.0 x 10-5 mmol cm-2 yr-1 for approximation via one vs. two lines, respectively. The sulfate profile for Site 1000 is provided in the "Carbonate Reactions" section.
In a general sense, the calculated rates of sulfate reduction (in mmol cm-2 yr-1), which range from 2.5 × 10-5 to 18.0 × 10-5 (Table 2), are low and consistent with the rates presented by Canfield (1991) for deep-sea sediments along the margins of ocean basins. This fundamental conclusion is insensitive to any reasonable manipulation of the calculation outlined above (e.g., choice of Ds or curve fitting the sulfate data). By contrast, the organic-rich surface sediments of the anoxic Cariaco Basin (Shipboard Scientific Party, 1997d) show a tenfold decrease in dissolved sulfate concentration over only 6 m of burial depth (see Lyons et al., 1998). Although the rates presented in Table 2 are only estimates, they do show a general positive relationship with respect to TOC concentration over the narrow ranges for both parameters. Sulfate depletion at Site 1000 is further complicated by the possibility of gaseous hydrocarbons providing additional electron donors to facilitate bacterial sulfate reduction. Details for Site 1000 are provided below in the "Carbonate Reactions" discussion.
The low TOC values and
correspondingly low rates of sulfate reduction are ultimately a function of
generally low surface primary production away from continental margins (coastal
upwelling) and the great water depths and slow rates of sediment accumulation at
the Leg 165 sites. Suess (1980) and others have demonstrated a strong inverse
relationship between water depth and the percent of surface organic-carbon
production preserved, reflecting progressive degradation within the settling
particulates. Furthermore, it is well established that carbon burial efficiency
(i.e., the proportion of the flux of organic carbon to the sediment surface that
survives diagenetic oxidation and becomes permanently buried) is positively
related to sedimentation rate (Müller and Suess, 1979; Henrichs and Reeburgh,
1987; Canfield, 1989b, 1994). Rapid burial not only fosters enhanced
preservation but also increases the availability of reactive organic phases for
sulfate reduction (i.e., favors enhanced survival from aerobic respiration in
the uppermost layers). This relationship manifests as a positive correlation
between the rate constant for sulfate reduction (k) and sedimentation rate (![]() )
(Toth and Lerman, 1977; Berner, 1978, 1980), which can be stated as
)
(Toth and Lerman, 1977; Berner, 1978, 1980), which can be stated as
where ![]() is an empirical constant. Consequently, the comparatively low rates of
accumulation at the pelagic to hemipelagic sites of Leg 165 are consistent with
both low rates of sulfate reduction and pore-water profiles bearing strong
signals of suboxic redox processes.
is an empirical constant. Consequently, the comparatively low rates of
accumulation at the pelagic to hemipelagic sites of Leg 165 are consistent with
both low rates of sulfate reduction and pore-water profiles bearing strong
signals of suboxic redox processes.
Given the low rates of
sulfate reduction summarized in Table 2
and the aforementioned strength of the suboxic signal, it is not surprising to
see low total sulfur contents in the sediments, with mean values of ![]() 0.16
wt% (Table 2). Despite the
availability of iron, inadequate supplies of bacterially produced H2S
limit the amount of sedimentary pyrite formation. In other words, detritally
delivered reactive iron is in sufficient supply despite the high dilution by
calcium carbonate (Berner, 1984; Canfield, 1989a; Canfield et al., 1992; Lyons
and Berner, 1992; Raiswell and Canfield, 1998). Overall, total sulfur
concentrations, which reflect pyrite in these sediments, are generally low and
nonsystematic in the sediments at the four sites (e.g., Fig.
3).
0.16
wt% (Table 2). Despite the
availability of iron, inadequate supplies of bacterially produced H2S
limit the amount of sedimentary pyrite formation. In other words, detritally
delivered reactive iron is in sufficient supply despite the high dilution by
calcium carbonate (Berner, 1984; Canfield, 1989a; Canfield et al., 1992; Lyons
and Berner, 1992; Raiswell and Canfield, 1998). Overall, total sulfur
concentrations, which reflect pyrite in these sediments, are generally low and
nonsystematic in the sediments at the four sites (e.g., Fig.
3).
Notable exceptions to the low pyrite sulfur contents are several of the discrete volcanic ash layers within the cores where pyrite enrichments, as secondary overgrowths on ash fragments, were first observed on board and later substantiated via the chromium reduction method described above (Table 3). It should be noted that these "discrete" layers, in many cases, bear bioturbational overprints of varying degrees. In addition to elevated sulfur contents, the ashes also show trace-metal concentrations that are enriched, particularly for nickel, relative to the host sediments and expected concentrations given the lithologies of the ash layers (Table 3) (Sigurdsson, Leckie, Acton, et al., 1997). This relationship is particularly well expressed in the ash layers from Site 999 where there is a general correlation between nickel and sulfur contents (Fig. 4). Furthermore, Site 999 shows unusually nonsystematic variation in pyrite sulfur content within the routinely analyzed bulk sediment samples (Fig. 3). This distribution of sulfur is reminiscent of the downcore variability that characterizes the distribution of ash within the core.
Preliminary evaluations suggest that secondary nickel and sulfur enrichments may be linked mechanistically to externally derived fluids bearing sulfide and metals that follow permeability pathways controlled by ash distribution. It is important to note that the pyrite enrichments do not reflect local sites of enhanced sulfate reduction (i.e., organic enrichment) or iron availability (iron is not limiting throughout the host sediments). Thus, it is difficult to envision the establishment of concentration gradients toward the ash layers. It is also important to note that this phenomenon is best expressed at Site 999 where high-angle faulting has been described in detail (Shipboard Scientific Party, 1997a). The sulfur isotope datawith the striking presence of 34S-depleted valuesare unambiguous in delineating a bacterial source for the sulfide (Ohmoto, 1972; Chambers and Trudinger, 1979; Machel et al., 1995; Lyons, 1997; Raiswell, 1997), which precludes basement-derived magmatic fluids or thermochemical sulfate reduction as a source for the sulfur (Table 3). The suggestion, therefore, is that bacterial sulfate reduction is occurring away from the sites of mineralization, perhaps deep within the sediment package, and that reducing fluids may be moving up along fault conduits and mineralizing the ash layers and, to a lesser extent, the adjacent host sediments. Such a model could also explain the atypical sulfur distribution in Figure 3. At this juncture we cannot preclude a basement origin for the metals, and the mechanistic details of metal and sulfur transport and emplacement, including the timing, remain uncertain.
![]()