-
(CH2O)106(NH3)16(H3PO4) + 53 SO42– +14 H+
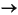
106 H2O + 106 CO2 + 16 NH4+ + HPO42– + 53 HS–. (2)
From the downhole variation of dissolved sulfate concentrations in the interstitial waters it is evident that bacterial sulfate reduction takes place in the sediment column of all sites investigated during Leg 201, with the exception of the open-ocean deep-sea Site 1231 (Fig. F2). The high-resolution sulfate measurements carried out during Leg 201 agree remarkably well with the less-resolved data obtained during the previous Leg 112 (Fig. F2). MSR associated with the degradation of organic matter leads to the liberation of carbon dioxide, ammonium, and hydrogen sulfide (e.g., Froelich et al., 1979) according to the overall reaction
Additionally, bicarbonate and bisulfide are produced upon sulfate reduction associated with anaerobic oxidation of methane (e.g., Hoehler et al., 1994; Boetius et al., 2000). By these two microbially catalyzed processes, sulfate reduction in Leg 201 cores is associated with an increase in pore water concentrations, such as dissolved sulfide, alkalinity, and ammonium (D'Hondt, Jørgensen, Miller, et al., 2003; D'Hondt et al., 2004). The intensity of net sulfate reduction, however, differs among sampling sites depending on the availability of organic substrates for microbial activity and sediment age. Thus, very low total organic carbon (TOC) contents characterize the sediments of Site 1231, which corresponds to Leg 34 Site 321 (Table T1).
Essentially constant sulfate concentrations with depth at the open-ocean Site 1231 (Fig. F2) (see also Brady and Gieskes, 1976, for preliminary results from Site 321) indicate that no net sulfate reduction occurs. This is confirmed by the sulfur isotopic composition of dissolved sulfate throughout the whole sediment column (see below) and is in agreement with results from oxygen isotope analysis of dissolved sulfate (Blake et al., this volume). Some net sulfate reduction is, however, observed at the equatorial deep-sea Sites 1225 and 1226, which receive a higher flux of organic matter because of enhanced productivity (D'Hondt, Jørgensen, Miller, et al., 2003). Sulfate concentrations increase toward the bottom of the sediment column following an initial decrease to a minimum value at ~200 meters below seafloor (mbsf). This increase suggests diffusion of sulfate from seawater that is flowing through underlying basaltic crust (D'Hondt, Jørgensen, Miller, et al., 2003). In contrast to the deep-sea sites, sulfate is completely exhausted at the Peru margin sites within the upper 30–40 mbsf (Fig. F2) because of higher amounts of reactive organic matter and enhanced sedimentation rates, which lead to the burial of a higher relative fraction of organic matter that may be easily consumed by microorganisms (Suess, 1980; Berner, 1980).
The experimental measurements of sulfate in the upper sulfate zone of Sites 1226–1229 gave depth-integrated gross sulfate reduction rates between 14 and 681 mmol/m2/yr (J. Kallmeyer and T.G. Ferdelman, pers. comm., 2004) (Table T3). These values indicate a dependence of the sulfate reduction rate on water depth, bottom water redox conditions, and, therefore, availability of organic matter. Sulfate reduction is typically less significant in the deep sea as compared to Peru margin sediments (Jørgensen, 1982; Canfield, 1991). The measured rates are lower than those typically measured on the continental margin (Jørgensen, 1982; Ferdelman et al., 1999; Fossing et al., 2000; Parkes et al., 2005; J. Kallmeyer and T.G. Ferdelman, pers. comm., 2004), but are higher than the modeled net sulfate reduction (e.g., Fossing et al., 2000; Böttcher et al., 2004b). The latter may be due to the fact that the activity of sulfate-reducing bacteria as deduced from pore water sulfate profiles tends to underestimate gross rates. Because of reoxidation of sulfide and enhanced transport processes in marine surface sediments, gross sulfate reduction rates there are typically much higher than the net rates reflected by dissolved sulfate (e.g., Fossing et al., 2000). Reduction of dissolved sulfate in the sediment is linked with the availability of metabolizable organic matter or hydrocarbons (mainly CH4) (Berner 1980; Borowski et al., 1996; Boetius et al., 2000). The sulfate profiles at Peru margin Sites 1227, 1228, and 1229 show a convex-upward curvature (Fig. F2), which, together with the downhole variations in alkalinity and dissolved ammonium (D'Hondt, Jørgensen, Miller, et al., 2003), indicate that sulfate reduction associated with in situ microbial degradation of organic matter plays a significant role in the upper part of the sedimentary column at these sites (Borowski et al., 1996). Downcore changes in sulfate gradients are observed at Sites 1228 and 1229 and suggest a nonsteady-state contribution of sulfate reduction, probably due to changes at the beginning of the Holocene. The different sulfate gradients seem to be related to changes in sedimentation rates and corresponding changes in lithology (D'Hondt, Jørgensen, Miller, et al., 2003). In the different lithologic units of the Peru margin sites, changes in sedimentation rates (Table T1) are associated with different organic matter contents, leading to changes in substrate availabilities. High methane concentrations were not observed at Sites 1225 and 1226, due to the availability of dissolved sulfate throughout the sediment core. Therefore, no contribution from anaerobic oxidation of methane is expected at these sites.
The saline brines found at depth at Sites 1228 and 1229 are enriched in sulfate compared to modern seawater (as much as 40 mM) (Fig. F2) but still have isotope signatures that exceed the modern seawater value. Based on the composition of the fluids it is assumed that seawater was the source of sulfate and that microbial sulfate reduction already modified these brines in their sulfate contents and associated stable isotopic composition.
Microbial reduction of dissolved sulfate causes a kinetic isotope effect and an enrichment of the lighter sulfur isotope 32S in the formed hydrogen sulfide (Kaplan and Rittenberg, 1964; Chambers and Trudinger, 1979). Both open- and closed-system diagenesis lead to a corresponding accumulation of the heavy isotope (34S) in the residual sulfate (Hartmann and Nielsen, 1969; Jørgensen, 1979; Jørgensen et al., 2004). Dissolved sulfate in the modern ocean has a sulfur isotopic composition of ~21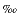 (Böttcher et al., 2000b), which does not differ significantly from the seawater composition within the past 50 m.y. (Paytan et al., 1998). This value is closely reflected by dissolved sulfate throughout the complete pore water profile at Site 1231, indicating that no net sulfate reduction takes place. The very low activity of sulfate-reducing bacteria parallels the very low TOC contents (Table T1). Pore water constituents, however, indicate some suboxic diagenesis (D'Hondt et al., 2004; D'Hondt, Jørgensen, Miller, et al., 2003). All other investigated sites showed a downcore increase in pore water sulfate 34S/32S ratios, mirroring the net decrease in dissolved sulfate (Fig. F2) and indicating that the changes in sulfate concentrations are because of bacterial activity. Isotope results obtained at Site 1228 are in general agreement with previous measurements carried out on sulfate at Site 680 (Fig. F2) (G. Claypool, unpubl. data). The maximum isotope values observed in the residual dissolved sulfate decrease with increasing water depth. This trend corresponds to a decrease in the overall activity of the sulfate reducing community (Table T3).
(Böttcher et al., 2000b), which does not differ significantly from the seawater composition within the past 50 m.y. (Paytan et al., 1998). This value is closely reflected by dissolved sulfate throughout the complete pore water profile at Site 1231, indicating that no net sulfate reduction takes place. The very low activity of sulfate-reducing bacteria parallels the very low TOC contents (Table T1). Pore water constituents, however, indicate some suboxic diagenesis (D'Hondt et al., 2004; D'Hondt, Jørgensen, Miller, et al., 2003). All other investigated sites showed a downcore increase in pore water sulfate 34S/32S ratios, mirroring the net decrease in dissolved sulfate (Fig. F2) and indicating that the changes in sulfate concentrations are because of bacterial activity. Isotope results obtained at Site 1228 are in general agreement with previous measurements carried out on sulfate at Site 680 (Fig. F2) (G. Claypool, unpubl. data). The maximum isotope values observed in the residual dissolved sulfate decrease with increasing water depth. This trend corresponds to a decrease in the overall activity of the sulfate reducing community (Table T3).
Minimum stable sulfur isotope fractionation during MSR was estimated in a first step using a (closed system) Rayleigh fractionation model (Goldhaber and Kaplan, 1974; Claypool, 2004). Measured sulfate concentrations [SO4] and isotope ratios (R) were modeled according to the following equation:
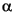 R–1) log [SO4] + log(R°) – (1 – R –1) log [SO4]°, (3)
R–1) log [SO4] + log(R°) – (1 – R –1) log [SO4]°, (3)
where
° = conditions at the sediment/water interface and
R = the fractionation factor.
In agreement with Equation 3, the pore water data gave essentially straight lines in a Rayleigh plot (Fig. F3). The resulting R values obtained from linear regression of the data at each site are between 1.014 and 1.040 (Table T3). These results are in the range previously calculated for marine sediments (e.g., Hartmann and Nielsen, 1969; Rudnicki et al., 2001; Böttcher et al., 2004b; Claypool, 2004). Because of the influence of transport processes (as diffusion) on pore water profiles, the actual in situ isotope fractionation factors are typically underestimated by the application of Equation 3 (Jørgensen, 1979; Goldhaber and Kaplan, 1980; Rudnicki et al., 2001; Jørgensen et al., 2004). For example, for the span of R values compiled in Table T3, model calculations of Rudnicki et al. (2001) for deep-ocean sediments of the Cascadia Basin gave actual isotope fractionations as much as 10 higher than closed-system simulations. These magnitudes of sulfur isotope discrimination obtained from pore water sulfate are within the range observed in experiments with pure cultures of mesophilic and psychrophilic sulfate-reducing bacteria, where a maximum of –47 has been observed (Kaplan and Rittenberg, 1964; Chambers et al., 1975; Canfield, 2001; Bolliger et al., 2001; Brüchert et al., 2001). It has additionally been found that sulfur isotope discrimination associated with anaerobic methane oxidation (Böttcher et al., 2000a) is similar to isotope effects using other electron donors (Kaplan and Rittenberg, 1964; Chambers and Trudinger, 1979; Detmers et al., 2001). Depending on the actual cellular rates of sulfate reduction (D'Hondt et al., 2002), isotope discrimination associated with this process may change (Kaplan and Rittenberg, 1964; Chambers and Trudinger, 1979; Chambers et al., 1975) and reactions from the oxidative part of the sulfur cycle may contribute to the overall isotope effects (Habicht and Canfield, 2001). Hydrogen sulfide may be reoxidized to sulfur species with intermediate oxidation states, such as elemental sulfur or thiosulfate. It has been found that the bacterial disproportionation of sulfur intermediates is associated with the production of 32S-enriched sulfide (Canfield and Thamdrup, 1994; Canfield et al., 1998; Cypionka et al., 1998; Habicht et al., 1998). It should be noted, however, that in deep hypersulfidic sediments from the Australian continental margin (Wortmann et al., 2001) and in deep-sea sediments of the Cascadia Basin (Rudnicki et al., 2001), sulfur isotope discrimination even exceeding –70 during bacterial reduction of dissolved sulfate was related to the metabolic activity of natural bacterial populations without implying further recycling by sulfur intermediates.
Claypool (2004) used results from a number of previous DSDP and ODP pore waters to derive a relationship between the openness of a sediment with respect to diffusional supply of dissolved sulfate and the calculated Rayleigh isotope fractionation factors. Results on modeled isotope discrimination from the present study (Table T3) together with recent published data from the southwest Pacific (Böttcher et al., 2004b) have been converted by the use of the relationship proposed by Claypool (2004) to derive the fraction between the diffusional flux (Jd) and the total flux (diffusional and burial) of sulfate (Jd + Jb). Results are compiled in Table T3 and indicate that all sediments considered here are at least partially open with respect to dissolved sulfate from the bottom waters with a minimum value for the fraction Jd/(Jd + Jb) in Leg 201 sediments of 0.46 (Table T3). Results range up to an 82% contribution of the diffusional flux and cover the range estimated for Leg 181 sediments from the southwest Pacific (Table T3).
Sedimentary reduced sulfur as a product of dissimilatory sulfate reduction was found in the Peru margin sites and is essentially present as pyrite with minor contributions from organic sulfur (Emeis and Morse, 1990; Mossmann et al., 1990). However, sedimentary sulfur may also occur in oxidized form as biogenic barite (e.g., Paytan et al., 1998), which is used as a proxy for paleoproductivity (Paytan et al., 1996) and typically contains the isotope signature of sulfate from the water column or in surface sediments (Paytan et al., 1998). In the zone of complete sulfate depletion, biogenic barite may redissolve and barium may diffuse upward in the sediment column toward the lower part of the sulfate zone. Because of the low solubility of barium sulfate, barite may precipitate at a "diagenetic front" (Brumsack et al., 1992; Torres et al., 1996; Böttcher et al., 2003; D'Hondt, Jørgensen, Miller, et al., 2003). Based on the pore water profiles observed in the sediments recovered during Leg 201 (Fig. F2) (D'Hondt, Jørgensen, Miller, et al., 2003), the dissolution of biogenic barite and the development of a diagenetic barite front can be inferred at Sites 1227, 1229, and probably 1228 (Fig. F2). The position of the barite front is expected to be at 30–40, 40, and 30 mbsf at Sites 1227, 1228, and 1229, respectively. The occurrence of a sulfate-rich brine at depth at Sites 1228 and 1229 can lead to the parallel development of a second, deeper-seated barite front at ~50 (Site 1228) and ~90 (Site 1229) mbsf. Based on the present isotopic composition of dissolved sulfate (~80–90 V-CDT), the diagenetic barite should be highly enriched in 34S with respect to modern biogenic barite (Paytan et al., 1998) and Pacific Ocean water sulfate (Longinelli, 1989; Böttcher et al., 2000a, 2004b). As introduced by Böttcher et al. (2004a), the magnitude of heavy (sulfur and oxygen) stable isotope enrichments in diagenetic barite may serve as an indicator for the steepness of paleosulfate gradients during the development of barite fronts. Investigations in cooperation with Glen Snyder have been initiated to look at the stable sulfur isotopic composition of barite in the diagenetic fronts that developed in sulfate-depleted Leg 201 sediments. At Sites 1225, 1226, and 1231, interstitial water sulfate does not decrease to an extent that biogenic barite remobilization and formation of diagenetic barite is expected.
![]()