GEOCHEMISTRY
Interstitial Water Sampling and Chemical Analyses
Shipboard interstitial water analyses were performed on 5- to 10-cm-long whole-round sections from Section 2 of each core. Samples were cut and capped immediately after core retrieval on deck. After extrusion from the core liner in the chemistry laboratory, the surface of each whole-round section was trimmed with a spatula to remove potential contamination related to coring. When whole-round samples were collected more quickly than they could be squeezed, the samples were capped on both ends and stored in the refrigerator for up to several hours.
Interstitial water was collected using the trace metal noncontaminating titanium squeezer, modified after the standard ODP stainless steel squeezer of Manheim and Sayles (1974). Pressure up to 205 MPa (30,000 psi) was applied using a hydraulic press. Interstitial water was passed through a prewashed Whatman number 50 filter fitted above a titanium screen and subsequently extruded into a plastic syringe attached to the bottom of the squeezer assembly. All interstitial water samples were double-filtered through 0.45-µm sterile Acrodisc filters. Aliquots for future shore-based analyses of strontium isotopes were placed in polytubes and heat-sealed. Aliquots for sulfur isotopes were placed in polytubes then poisoned with cadmium acetate solution (1 mL of 1-M Cd[C2H3O2]2 to 10 mL of interstitial water sample) and heat-sealed. Aliquots for future shore-based analyses of interstitial water 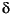 13C were poisoned with powdered mercuric chloride (~3-5 mg to 4.5 mL of interstitial water sample), and aliquots for future shore-based analysis of trace metals were acidified with concentrated nitric acid (100 µL of HNO3 to 4 mL of interstitial water sample). Both were placed in individual glass vials and double-sealed with parafilm.
13C were poisoned with powdered mercuric chloride (~3-5 mg to 4.5 mL of interstitial water sample), and aliquots for future shore-based analysis of trace metals were acidified with concentrated nitric acid (100 µL of HNO3 to 4 mL of interstitial water sample). Both were placed in individual glass vials and double-sealed with parafilm.
Interstitial water was routinely analyzed for salinity and total dissolved solids with a Goldberg optical handheld refractometer (Reichert) and for pH and alkalinity by Gran titration with a Brinkmann pH electrode and a Metrohm autotitrator. Chloride concentrations were determined by titration with AgNO3. Silica, phosphate, and ammonium determinations were carried out by colorimetry using a Milton Roy Spectronic spectrophotometer using the analytical techniques described by Gieskes et al. (1991).
Sulfate was analyzed by ion chromatography (ICr) using the Dionex DX 120 ion chromotagraph. Potassium, calcium, and magnesium concentrations were not determined by ICr because of an unidentified problem that persisted even after changing the cation column and the suppressor. These elements were analyzed by ICP-AES.
The major and minor cation concentrations (K, Ca, Mg, Na, Li, B, Sr, Ba, Mn, and Fe) were determined using the Jobin-Yvon Ultrace ICP-AES following the procedure outlined by Murray et al. (2000). In preparation for analysis by ICP-AES, 10-mL aliquots of interstitial water were acidified with 10 mL of nitric acid (HNO3) and diluted tenfold with matrix solution (2.25% HNO3 containing 9 ppm Y) for minor elements and diluted fiftyfold for major elements. Analytical blanks were prepared in an identical manner by analyzing nanopure water acidified and diluted with matrix solution to ensure a matrix match with the interstitial water samples. Sodium was determined using a charge balance calculation, where 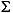 cations = anions.
cations = anions.
Dissolved organic carbon concentrations were measured using a TOC-5000A analyzer. After interstitial water was extracted from the squeeze cake, ~1.5 mL of the water was removed and quickly frozen to preserve the sample until an analysis could be made. Samples were diluted fivefold (1 mL of sample to 5 mL of nanopure water) and acidified to a pH of ~2 using 53 mL of 2-N hydrochloric acid (HCl). Samples were then purged with purified air for 3 min (50 mL/min) and analyzed by triple injections of 25 µL of sample.
International Association of Physical Sciences of Organization (IAPSO) standard seawater was used for calibrating most techniques. The reproducibility of these analyses, expressed as a percent of the standard deviation (1 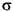 ) divided by the average of several IAPSO values, is summarized in Table T10. Accuracy of individual analysis is within the accepted deviation range for the accepted IAPSO values.
) divided by the average of several IAPSO values, is summarized in Table T10. Accuracy of individual analysis is within the accepted deviation range for the accepted IAPSO values.
Bulk Sediment Sampling and Chemical Analyses
Bulk sediment samples, taken from interstitial water squeeze cakes, were routinely analyzed during Leg 206. Approximately 50 g of sample was ground in an agate mortar to a fine powder after freeze-drying. Sediment samples were analyzed for calcium carbonate using a Coulometrics 5011 carbon dioxide coulometer and for total carbon using a Carlo Erba 1500 CNS analyzer. Organic carbon was calculated from the difference between total carbon and inorganic carbon.
Elemental analyses for bulk sediment samples were measured by ICP-AES as outlined by Murray et al. (2000). Samples and standards (0.1000 ± 0.0002 g) were mixed with lithium metaborate (LiBO2) flux (0.4000 ± 0.0004 g). Analytical blanks were prepared with 0.4000 ± 0.0004 g LiBO2 flux to ensure matrix matching. A solution of 0.172-mM LiBr wetting agent (10 mL) was added to the samples, standards, and blanks to prevent the cooled bead from sticking to the sides of the crucible. This mixture was fused for 3 min at 900°C in a NT-2100 Bead Sampler prior to dissolution in 50 mL of 10% HNO3. For complete dissolution, 1 hr of shaking with the Burrell wrist-action shaker was required. A 5-mL aliquot of the resulting solution was filtered (0.20 µm) and diluted with 35 mL of 10% HNO3, resulting in a 4000-fold dilution of the original powder. Concentrations of the major elements (Si, Ti, Al, Fe, Mn, Mg, Ca, Na, K, and P) are presented as weight percent of oxides and trace elements (V, Cr, Sr, Y, Zr, and Ba) as parts per million (ppm). Phosphorous and zirconium were difficult to measure (analytical uncertainty 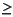 30%) and are not presented in the initial results.
30%) and are not presented in the initial results.
For these measurements, analyses of certified standards (BCSS-1 [marine mud], BHVO-2 [Hawaiian basalt], JCH-1 [Japanese chert], JLS-1 [Japanese limestone], MAG-1 [marine mud], and NIST-1C [argillaceous limestone]) were used to develop a six-point calibration curve for the major and trace elements. These standards were chosen because they are most representative of the lithologies at Site 1256. The results of BHVO-2 were poor and were taken out of the calibration. The drift was monitored with a synthetic solution composed of multiple natural samples and spikes of a few trace elements as needed. A certified standard (SCO-1 [Cody Shale]) was also analyzed as an unknown to check accuracy and consistency between analyses. The reproducibility of this analysis, expressed as a percent of the standard deviation (1 ) divided by the average of six (three times per run) determinations of an unknown sample, is summarized in Table T11. The analytical uncertainty for the bulk sediments is relatively high. Because of this and because the supply of argon was limited and intended for use with hard rock analyses, sediment analyses were only allotted a minimum number of runs. The quality of the analyses is, however, acceptable for observing general lithologic trends in the sediment column.
The sediment samples analyzed by ICP-AES were ignited before dissolution to release volatile phases (H2O, CO2, and SO2) and to fully oxidize all iron to ferric iron. The ICP-AES analyses of these ignited powders should total 100% if the dilution measurements are perfect, but this was not the case and sample totals were consistently below 100%. Inorganic carbon (CO32-) is independently measured by coulometry, and the amount of CaO required to balance the inorganic carbon should approximately match the concentration of CaO measured by ICP-AES. As this was also not the case, the assumption was made that all the calcium present in the sediments is present as calcium carbonate and that there are only trace concentrations of other elements (Mg, Sr, and Fe) in the carbonate. Following this assumption, the ICP-AES data were normalized such that the CaO concentrations were consistent with the amount required to balance the inorganic carbon determined by coulometry. The volatile components (H2O and CO2) were then combined with the ICP-AES analyses and the data normalized to 100 wt%. These normalized data are reported in "Inorganic
Geochemistry" in "The Sedimentary Overburden (Holes 1256A, 1256B, and 1256C)" in the "Site 1256"
chapter (see Table T23 in the "Site 1256" chapter).
Hard Rock Sampling and Geochemical Analyses
Representative samples from selected igneous units were analyzed for major and trace elements during Leg 206 using ICP-AES. Approximately 20-cm3 samples were cut from the core with a diamond saw blade. All outer surfaces were ground on a diamond-impregnated disk to remove surface contamination and altered rinds resulting from drilling. Each cleaned sample was placed in a beaker containing trace metal-grade methanol and was ultrasonicated for 15 min. The methanol was decanted, the samples were ultrasonicated twice in deionized water for 10 min, and then were ultrasonicated 10 min in nanopure DI water. The clean pieces were then dried for 10-12 hr at 65°C.
The clean, dry, whole-rock samples were fragmented to chips <1 cm by crushing them between two disks of Delrin plastic in a hydraulic press. They were then ground to a fine powder in a tungsten carbide (WC) mill by a SPEX 8510 shatterbox. A 1.0000 ± 0.0005-g aliquot of the sample powder was weighed on a ScienTech balance and ignited to determine weight loss on ignition (LOI).
ODP Technical Note 29 (Murray et al., 2000) describes in detail the shipboard procedure for dissolution of rocks and ICP-AES analysis of samples. The following protocol is an abbreviated form of this with minor changes and additions. After determination of LOI, 100.0 ± 0.2-mg aliquots of the ignited whole-rock powders were weighed and mixed with 400.0 ± 0.5 mg of LiBO2 flux that had been preweighed on shore. Standard rock powders and full procedural blanks (400 mg LiBO2) were included with the unknowns in each ICP-AES run. In addition, a grinding blank of pure SPEX SiO2 was included in Runs 1 and 2 as a check on grinding contamination contributed by the WC mills (Table T12). The grinding blank was processed using the shatterbox that appeared dirtiest and is therefore a "worst-case" scenario. All samples and standards were weighed to ±0.20 mg on the ScienTech balance, and weighing errors are estimated to be ~0.02 mg.
We added 10 mL of 0.172-mM aqueous LiBr solution to the flux and rock powder mixture as an antiwetting agent to prevent the cooled bead from sticking to the crucible. Samples were then individually fused in Pt-Au (95-5) crucibles for ~3 min at a maximum temperature of 1050°C in a Bead Sampler NT-2100. After cooling, beads were transferred to 125-mL high-density (HD) polypropylene bottles and dissolved in 50 mL 10% HNO3, aided by shaking with a Burrell wrist-action bottle shaker for 1 hr. From Run 2 onward, the samples were ultrasonicated for ~1 hr after shaking to ensure complete dissolution of the glass bead. After digestion of the glass bead, all of the solution was passed through a 0.45-µm filter into a clean 60-mL wide-mouth HD polypropylene bottle. Next, 2.5 mL of this solution was transferred to a plastic vial and diluted with 17.5 mL of 10% HNO3 to bring the total volume to 20 mL. The final solution-to-sample dilution factor for this procedure was ~4000-fold.
Major (Si, Ti, Al, Fe, Mn, Mg, Ca, Na, K, and P) and trace (Sc, V, Cr, Ni, Sr, Y, Zr, Nb, and Ba) element concentrations of standards and samples were determined with the JY2000 Ultrace ICP-AES, which routinely measures wavelengths between ~100 and 800 nm. Specific analytical conditions for each sample run during Leg 206 are provided in Table T13.
The JY2000 plasma was ignited at least 30 min before each sample run to allow the instrument to warm up and stabilize. After the warm-up period, a zero-order search was performed to check the mechanical zero of the diffraction grating. After the zero-order search, the mechanical step positions of emission lines were tuned by automatically searching with a 0.002-nm window across each emission peak using the BAS-148 standard (basalt standard created during Leg 148, Hole 504B; Bach et al., 1996), or the BAS-206 (basalt interlaboratory standard created during this leg) prepared in 10% HNO3. During the initial setup an emission profile was selected for each peak, using BAS-148, to determine peak-to-background intensities and to set the locations of background points for each element. The JY2000 software uses these background locations to calculate the net intensity for each emission line. The photomultiplier voltage was optimized by automatically adjusting the gain for each element using BAS-148.
ICP-AES data presented in "Hard Rock Geochemistry" in "Basement Formed at Superfast Spreading Rate (Holes 1256C and 1256D)" in the "Site 1256" chapter were acquired using either the Gaussian or Maximum mode of the Windows 5 JY2000 software. The Gaussian mode fits a curve to points across a peak and integrates the area under the curve to determine element intensity and was used for Si, Ti, Al, Fe, Ca, Na, K, Sc, V, Sr, Zr, and Nb. Maximum mode was used for elements with asymmetric emission peaks (Mn, Mg, P, Y, Cr, Ni, and Ba), and intensity is integrated using the maximum intensity detected. Each unknown sample was run at least twice, nonsequentially, within a given sample run.
A typical hard rock ICP-AES run (Table T14) during Leg 206 included (1) a set of five certified rock standards (JA-3, JB-3, JB-2, BHVO-2, and JGb-1) analyzed twice each throughout the sample run; (2) up to 20 unknown samples run in duplicate; (3) a drift-correcting sample, BCR-2; spiked with Ni after Run 2 and P after Run 3, analyzed every fourth sample position and at the beginning and end of each run; (4) blank solutions run near the beginning and end of each run; and (5) a check standard (i.e., standard run as an unknown), typically BAS-148 and BAS-206, although Run 2 consisted primarily of check standards. A 10% HNO3 wash solution was run for 90 s between each analysis.
Following each sample run, the raw intensities were transferred to a data file and all samples were corrected first for drift and then for the full procedural blank. The drift correction was applied to each element by linear interpolation between drift-monitoring solutions run approximately every fourth analysis. Following drift correction and blank subtraction, calibration curves were constructed based on five certified rock standards (JA-3, JB-3, BHVO-2, JB-2, and JGb-1 for Run 2 onward). Unknown concentrations were then calculated from the calibration line.
Estimates of accuracy and precision for major and trace element analyses were based on replicate analyses of check standards (usually BAS-148 and BAS-206), the results of which are presented in Table T15. In general, run-to-run relative precision by ICP-AES was <3.5% for the major elements. Run-to-run relative precision for trace elements was generally <9%. Exceptions typically occurred when the element in question was near background values.
